Could Aging be an Infectious Disease?
Making the case that aging could be primarily driven by infections, told from the perspective of a future “Mick and Rorty” episode … for kicks

======= (TV Voice) Previously on Mick and Rorty ===========
Mick : Wow Rorty, it seems like a lot of the genes up-regulated in old-age, center around the complement pathway of the immune system
Rorty : What do you think it means Rick?
Mick : Not sure really, but I wouldn’t be surprised if a lot of disease is due to our bodies’ reaction to everyday pathogens. I mean, think about it! EEEEVERYONE gets the common cold and EEEEEVERYONE dies Rorty. In some parallel dimensions I’ve been to, they actually take the vaccination against infectious agents seriously and don’t bash vaccines. People live so long they die of boredom!
Rorty : Uh … I don’t know Rick …. I think you’re drunk again
Mick : I’m sober enough to get your non-infringing name right …… “Rorty”
========================================
Previously when we looked at the 2009 meta analysis by the Church lab of 27 different aging gene expression datasets, we found many genes linked to the complement cascade were enriched, along with several genes associated with mitochondrial function and Reactive Oxygen Species (ROS) cleanup. None of us should be surprised about the mitochondrial connections, but the complement pathway hyperactivation is interesting. Could this specific aspect of the immune system underlie aging in so many tissues? Is this the core of “inflammaging/immunosenescence”?
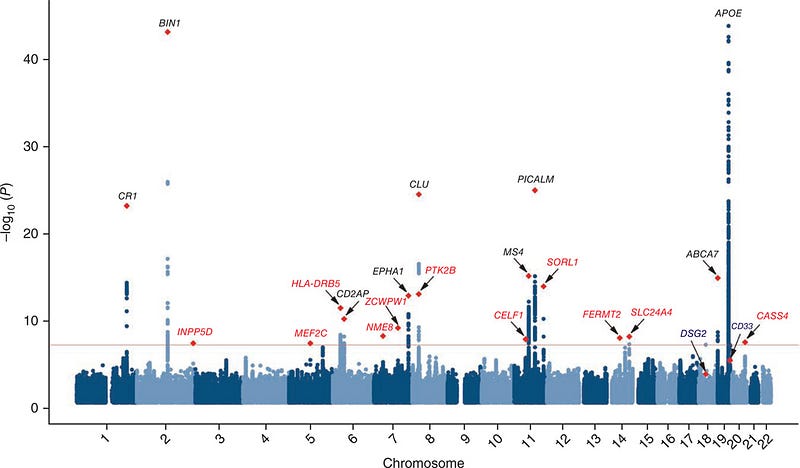
The overexpression of these genes seems to align with what is knwon about the immune system’s role in Alzheimer’s and cardiovascular disease. In Alzheimer’s, the CR1 (complement receptor 1) gene is a top GWAS candidate, and this receptor binds to complement C3, which is a gene highly expressed in aging tissues.
Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease
In heart disease, activation of the complement cascade appears to play a central role in the formation of atherosclerotic plaques, and incidentally many of the genes in the 2009 meta-analysis point to atherosclerosis pathways in the literature. However, we need to connect these pathways to the genomic GWAS data, particularly the primary associated locus. The plot below is a manhattan plot from the CARDIOGRAM study, and shows the top genes associated with risk of developing cardiovascular disease or an actual heart attack, which is ultimately responsible for 1/4 of all deaths in the US.
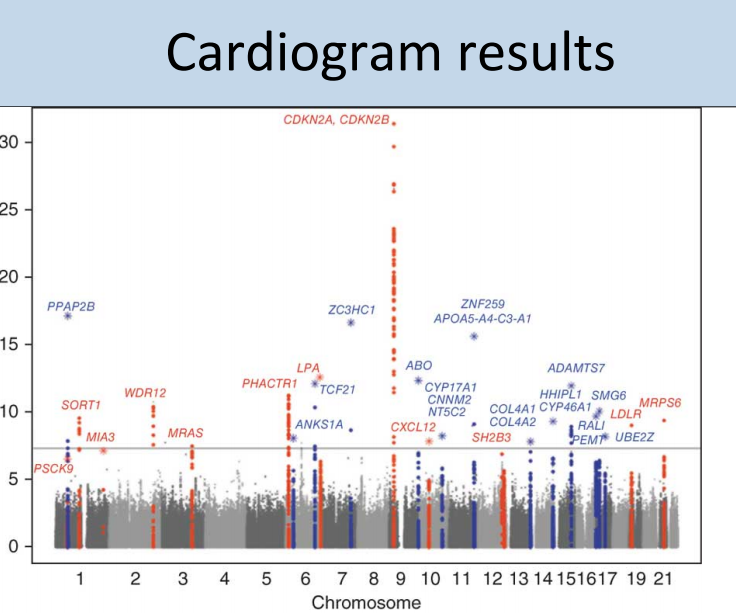
That big peak in the middle, CDKN2A, is p14/p16 …. which many of you will recognize as THE PRIMARY MARKER of cellular senescence, now a primary focus of aging research and longevity. This is neat, for evidently senescence of smooth muscle cells in blood vessels is associated with atherosclerosis. This is a clear indication that focusing on the biology of aging, directly, will yield tremendous benefits to medicine.
This is genomics GWAS data, which tells us how the genes we are born with influence disease risk. The dataset we previously dug into, however, told us which genes were turned up the most in aging tissues. Do you think we can connect this specific gene, and its genomic variants, directly to the previous gene expression data? We would need to find connections to the complement cascade and mitochondrial function. Well, we sure can! Meet my friend p32, an obscure protein that will be the center of this story. Hi P32!
This gene was independently discovered and characterized in several different labs, and given many different names :
SF2p32 — So named due to its association with pre-mRNA-splicing factor SF2, which regulates p14 and p16 splicing, among the alternate splicing of many other targets.
HABP1 : Hyaluronan binding protein 1 — which is interesting because elevated hyaluronan is suggested to be the longevity-promoting factor in naked mole rats
C1QBP: Complement 1Q binding protein — this gene has been shown to bind and inhibit complement C1Q, 3 genes of which are present in the aging dataset (in addition to complement C3 and C4A). C1QBP has also been called gC1qR because it is found as a membrane cell-surface receptor.
From here on out however, lets just call SF2p32/C1QBP/HABP1/ p32!
P32, turns out, has profoundly important roles in the mitochondria :
- p32 has a crucial role for regulating mitochondrial function by regulating the translation of mitochondrial proteins
- If you overexpress p32, you get increased mito function and reactive oxygen species (ROS). Knock it down, and cells become significantly reduced in their mitochondrial oxphos capacity
- In fact, cells lacking p32 are predominantly glycolytic. P32 may in fact have an important role in the warburg effect in cancer.
- Also intriguingly, p32 is a component of the mitochondrial permeability transition pore complex. It inhibits the permeability transition, keeping the mitochondria alive even if it’s making reactive oxygen species that would otherwise trigger apoptosis. Its a mitochondrial keep alive signal! The activity of the permeability transition pore has been shown to be relevant in cardiovascular pathologies.
Most importantly, this p32 gene is directly regulated by the top genetic cause of heart disease. The CDKN2A gene encodes both p16 (senescence marker) and an entirely separate protein p14ARF (made in an alternate reading frame!). Furthermore, there exists a truncated version of p14 that is small and mitochondrial, so lets call it … smARF!

The smARF version of p14 has been shown to translocate into the mitochondria and bind to p32. This interaction between the senescence machinery and the mitochondria has been shown to be a crucial part of p53-independent autophagy and mitophagy, which protect against cancer.
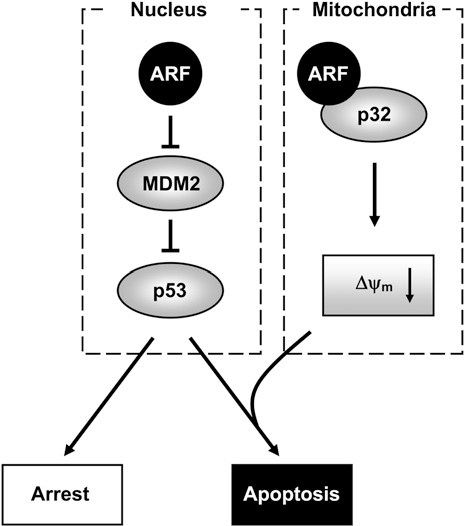
It gets even more exciting. P32 is found in the membrane of the cell, where it was first characterized for its ability to bind both complement 1Q and Hyaluronan. Some particularly exciting work has shown this cell-surface expression appears to be located specifically in hypoxic regions of tumors. Furthermore, p32 membrane expression correlates with tumor malignancy. This is exciting, for this may be a cell surface marker of mitochondrial health, or possibly even senescence.
=============== Intermission ==================
Rorty : Wow, gee-wiz Mick! That gene seems connected to a lot of stuff we care about in aging pathways…. mitochondria, ROS, the innate immune system…
Mick : I know Rorty, its like the mitochondria kills itself when its not needed anymore, but this thing keeps it alive juuuuuust long enough to power the cell if needed. The cell can keep a crappy mito around if it really needs the energy, even if its producing toxic chemicals.

Rorty : This reminds me of your car battery Mick …. with that whole tiny universe inside it, with those people you enslaved…
Mick : Rorty, you’re a genius! To keep that cellular car battery alive, you just need to get inside it and motivate it internally with the revelations that there is a whole outer universe that simultaneously depends on them but also doesn’t care about them! Its genius! The mitochondrion will never know the difference! That must be what p32 is doing. p14ARF on the other hand is ruining everything by going in there and telling the mitochondria it’s not needed anymore. We can’t let that happen!
I need my car!
========================================
Back to the science
So we have this gene (p32) that seems to intricately regulate mitochondrial fission/fusion, inhibits the permeability transition pore, potentiates autophagy/mitophagy, makes the cell resilient to oxidative stress (but leads to increases in ROS), and most importantly of all, directly binds to the p14ARF gene that is the top associated locus for heart disease (with its partner p16 being the top marker of cellular senescence). It also binds to, and sequesters complement C1Q, and the complement system overall had many genes upregulated in the prior meta analysis we focused on.
What now?
Recently, a paper came out that is getting people awfully excited. For whatever reason, Herpes Simplex Virus 1 (HSV1) is found in Alzheimer’s brains, and evidence for this has been mounting for years.
Here’s some highlights from an editorial that summarizes many prior studies:
- In mice, brain amyloid deposits are found after HSV1 infection
- Antiviral drugs block HSV1-induced A-beta and tau pathology
- In the brains of alzheimers patients, HSV1 DNA localizes with plaques!!!!
- Most recently, it was directly demonstrated that amyloid-beta, the primary feature of Alzheimer’s, surrounds herpesvirus to contain it and prevent it from spreading.
This is exciting, so I went digging to see if the same is true for cardiovascular disease, which kills 1/4 of all Americans. It is, and evidence has been mounting for decades that herpesvirus is causative.
- Having antibodies for HSV1 doubles your risk of heart disease
- Whats more, HSV1 and other viral infections seem to muck with cholesterol transport in affected cells, which is interesting because cholesterol has such a prominent role in both Alzheimer’s and heart disease. : “Evidence began accumulating >25 years ago suggesting that herpesviruses may possibly play a role in the development of atherosclerosis. It was found that Marek’s disease virus, an avian herpesvirus, caused typical atherosclerotic lesions in chickens, and when smooth muscle cells (SMCs) were infected with the virus in vitro, cholesterol accumulated”
And here’s the kicker. Viruses are clever little creeps. They evolve mechanisms to hijack our cells to increase the rate at which they can spread. It turns out that many viruses hijack the p32 protein to replicate.
- Herpes Simplex 1 (HSV-1) binds p32. If p32 is knocked down in the lab, there is a substantial reduction in the viral export from the cell.
- Mareks Disease Virus (MDV) is a Herpesvirus that infects chickens. It has been shown that MDV can directly cause atherosclerosis in chickens, however this does not happen if they are vaccinated prior to infection. Furthermore, evidence suggests that p32 is a binding partner of proteins that coat MDV.
- And its not just HSV-1! Epstein Barr Virus (EBV) is dependent on p32 for its replicative behavior : “The Epstein–Barr virus glycoprotein complex gMgN has been implicated in assembly and release of fully enveloped virus, although the precise role that it plays has not been elucidated. We report here that the long predicted cytoplasmic tail of gM is not required for complex formation and that it interacts with the cellular protein p32, which has been reported to be involved in nuclear egress of human cytomegalovirus and herpes simplex virus. “
- And its not just HSV-1 and Epstein Barr and Cytomegalovirus! Adenovirus too! “Adenovirus core protein V interacts with p32 — a protein which is associated with both the mitochondria and the nucleus.”
- And its not just HSV, Epstein Barr and Cytomegalovirus, and Adenovirus. EvenRSV (respiratory syncytial virus, causes many upper respiratory tract infections and kills more every year than influenza!) requires P32 binding for its reproduction : “Mitochondrial protein p32/HAPB1/gC1qR/C1qbp is required for efficient respiratory syncytial virus production” : “The results implicate p32 as a key host factor for RSV virus production, and bring to light the potential importance of mitochondria in RSV infection.”
- And if HSV, Epstein Barr, CMV, Adenovirus, and RSV weren’t enough, HIV viral replication is dependent on p32, as is replication of the rubella virus.
- Hepatitis C core protein binds to p32, inhibiting the T-lymphocyte response against the virus.
- Human Papilloma Virus (HPV) binds appears to bind p32, as does Ebola virus (EBV) through a mannose-binding protein mechanism.
- Finally, while there are no papers directly demonstrating that p32 is required for influenza replication, p32/c1qbp is one of a few genes associated with influenza severity in GWAS studies. This is highly suggestive that like the many other viruses in this list, influenza requires p32 to spread. This is relevant to heart disease, for it has been demonstrated that you are 6 times more likely to have a heart attack in the week following an influenza infection.
12 viral strains, so far (I’ll likely find more!), are dependent on this critical mitochondria and complement cascade modulating protein for survival. Why? Some work indicates that this may be relevant for export from the nucleus. Another possibility is exploiting p32’s ability to bind and sequester complement c1q to inhibit the immune response.
Many infectious agents appear to accelerate atherosclerosis in mouse models, particularly when APOE (the top Alzheimer’s gene)is knocked out. This tells us something very important about the role of cholesterol in the repair of infections in both alzheimer’s and cardiovascular disease.
This right here is quite fascinating, for it directly links a lot of plausible causal mechanisms of viral-mediated development of two major diseases. It also seems to link the aging gene expression data (complement cascades) with the top genetic causes of Cardiovascular and Alzheimer’s disease (Complement receptor, APOE, and p14/p16).
But why?
Why do viruses hijack this particular pathway? Why have so many different viruses, which have presumably evolved from different ancestors, all learned to attack the same critically vulnerable p32 protein? There are several things to consider here :
- Since p32 interacts with the immune system by forming a cell surface receptor and binding to complement C1Q proteins, these viruses may be using our own defense systems against us. Viruses are known to evade the complement pathway with a variety of other strategies.
- Modulating the mitochondria is critical to a virus. Since mitochondria can directly initiate cellular death, the virus must inhibit this pathway in order to keep the cell alive and replicate. Since p32 is part of this cell death pathway, it makes sense to exploit it.
- It goes deeper however ….
Since the p14ARF gene controls cell cycle, viruses may have an advantage in controlling that as well. When p14/p16 are overexpressed, the cell enters a state we call senescence, in which it freezes into a single state and stops dividing forever. As we all age, our cells accumulate senescent cells, which make us sicker. This is considered to be a dominant mechanism of aging.
It turns out that p14/p16 induces cell senescence by freezing the cell cycle precisely at the G1/S checkpoint. This is the end of the “growth phase” and precedes the “synthesis (S)” phase in which the genome in the nucleus is copied for cell division to occur later.
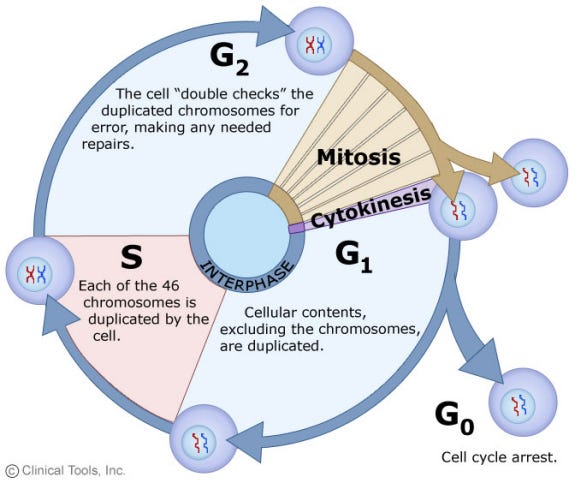
p14/p16, the gene that marks the state of senescence, specifically pauses the cell cycle at the transition between G1 and S. In cellular senescence, this pause becomes permanent. In healthy cells, this is a time for the cell to stop growing but prepare for the task of copying the entire genome, which requires energy. It is also at this state that mitochondria do something absolutely fascinating ….
Mitochondria, it turns out, aren’t always single isolated players. There are many thousands of mitochondria in the cell, however they are under phases of fission and fusion, in which they divide and recombine. Dr. Jason Fung has written about this phenomenon in an excellent post.
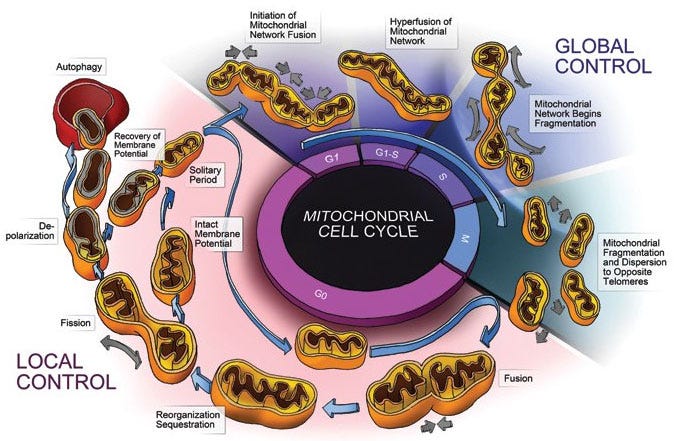
Miraculously, at the G1/S transition, mitochondrial combine into a single cell-wide network of connected powerhouses. At this state, the potential across the mitochondrial membrane increases dramatically, and ATP production is greatly enhanced, as is ROS.
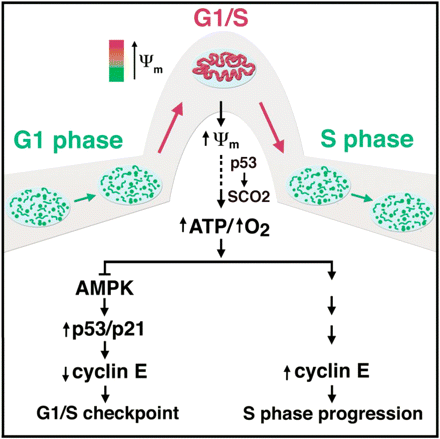
As it turns out, viruses have been found to interfere with this cell cycle in many ways. Some accelerate it, and others freeze it at specific locations. Many viruses freeze the cell cycle at this precise location to maximize viral replication. Its precisely at this state that the cell is producing abundant energy, and it is ready to copy the human genome, but it has not committed to doing so just yet. This is the perfect time to copy a viruses code instead.
There exists abundant evidence that this is happening :
- Herpes-Simplex-1, which we’ve previously discussed is linked to both Alzheimer’s and heart disease, is known to specifically arrest cells at the G1/S checkpoint.
- Vaccinia optimizes its regulation by disabling apoptosis by modulating mitochondria
- Cytomegalovirus enhances the expression of p14/p16, and this is required for optimal replication
- HPV, which causes cervical cancer, halts the cell at G1/S and G2/M checkpoints
- Hepatitis E promotes mitochondrial fusion to increase replication
- Many more examples are summarized in this excellent review
Most interestingly of all, cells in the senescent state are what we traditionally think of as a primary marker of aging. Senescence is specifically characterized by cells arrested in the G1/S state. These cells produce high levels of reactive oxygen species. These hallmarks of aging MATCH the hallmarks of viral replication, hijacking our cells to become factories for malicious code we do not want.
I mean, heck! It really does seem like this is all converging doesn’t it? If we can crack some of these pathways, just imagine how many more seasons we can expect!
=======================================
100 years more Rorty! If we cure heart disease and Alzheimer’s, we can go on more adventures. More and more Rorty! Forever and ever Rorty!