Decoding the Underlying Meaning of Aging Clocks
Much of this blog post was inspired by the wonderful conversations at the recent Foresight Longevity Frontiers conference! I am forever in their debt for inspiring this analysis.
The desire to “live forever” is as old as mankind, with several stories of historical figures attempting to escape death. 2,000 years ago the Chinese emperor “Wan Hu” strapped some of the earliest fireworks under his chair and arguably become the first astronaut in an attempt to escape to the immortal heavens. The Egyptian pharaohs also dreamed of a transition to an immortal afterlife, and thus built the great pyramids of Giza in order to facilitate that transition. I like to joke that this may have been the first ever “pyramid scheme”, although ChatGPT tells me the comparison is wrong. (buzzkill)
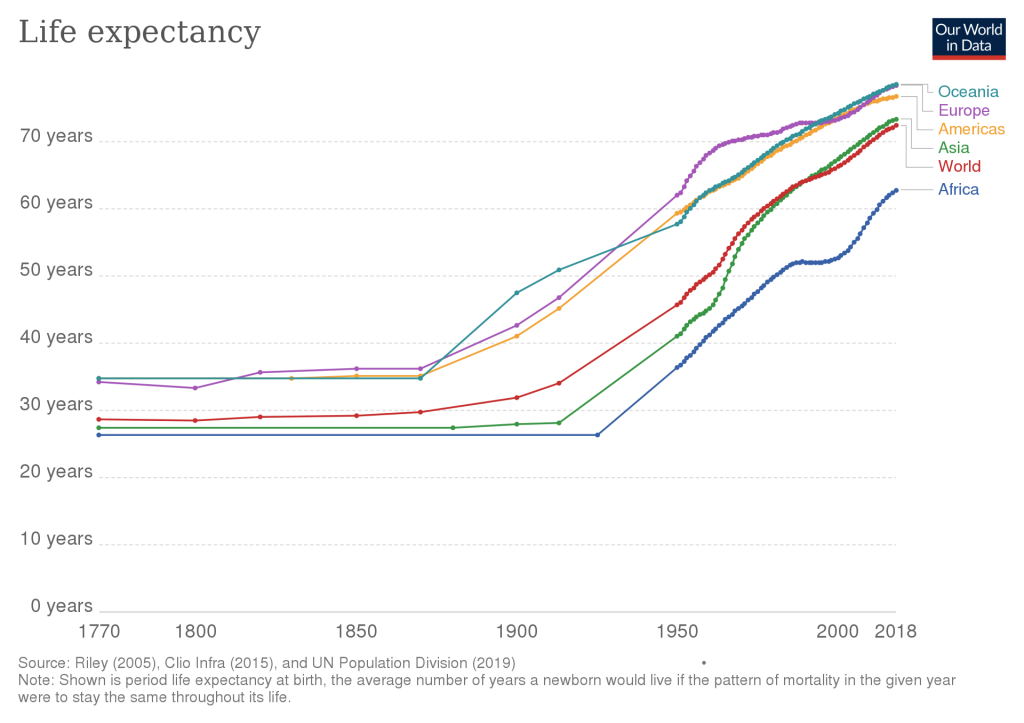
Those attempts at immortality in antiquity were clearly unsuccessful. In the 20th century however, mankind’s quest for longevity finally began to bear fruit. In the last 100 years, we have roughly doubled the human life expectancy, a notable acceleration in progress that has never been seen in millions of years of our species evolution. This should make you optimistic. If you are reading this, you have been born in a truly special time in our species history!
Now that we are in the 21st century, a new revolution has just started to map the underlying causes of aging itself. What will follow, moving forward, are unifying theories that link the slow progress of biological aging with the long-term development of disease. What a time to be alive!
Diet, Metabolism, and Human Longevity
With the sudden jump in lifespan over the last century, we are now seeing diseases of old age dominate as the top causes of death. Outside of the exciting longevity research field, more traditional clinical research has continued to slowly address the rising epidemic of metabolic syndrome and its associated sequelae : diabetes and obesity. These conditions underlie several diseases of aging, particularly heart disease, which kills the most people in modern countries. Obesity, in particular, is one of the strongest and earliest recognized biomarkers for predicting mortality. All of these diseases are associated with age, but can we find underlying mechanisms that tie them to aging itself? That’s what this post is all about.
Caloric Restriction
One of the oldest and most reproducible longevity interventions in lab animals is caloric restriction (CR). Some of the earliest work goes back to 1935, when it was shown that CR can extend the lifespan of rats. It is overly-simplistic to assert that caloric restriction and obesity are on opposite ends of a continuum, yet it is clear that there is a connection here, of some sort, between diet, obesity, and longevity. At the very least, as one of the first documented longevity interventions, it makes sense why our healthcare system has focused so much on body weight as a preventative measure.
Methionine Restriction
In the 1990s, a new discovery was made that refined our understanding of calories and aging. In fact, the calories may not have had anything to do with it at all! Amazingly, there is a single essential constituent of our diets that, when restricted, produces many of the same benefits to health and longevity as caloric restriction. That molecule is methionine, one of the 9 essential amino acids (out of 20) that our bodies cannot synthesize from scratch, but are the building blocks our cells need to make all proteins.
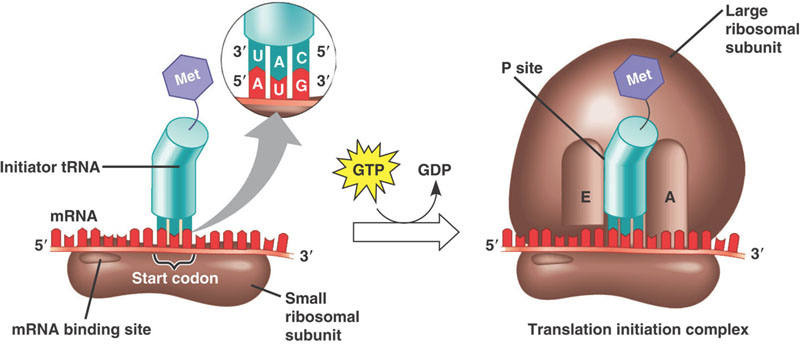
Methionine is also special in its own unique way, in that it is the first amino acid for every single protein your cells can make. It is the amino acid encoded by the start codon in every RNA transcript. Thus, without methionine, your cells can not even start to make any proteins.
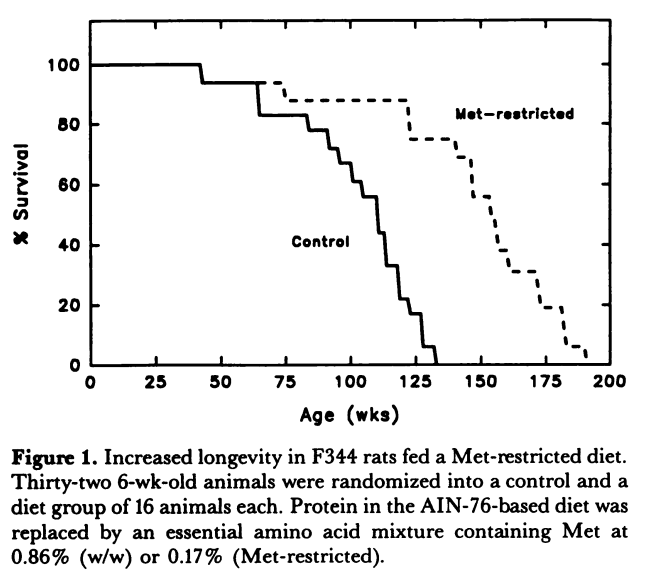
Methionine restriction was first shown in 1994 to extend the lifespan of rats by 30%. Since this landmark study, similar lifespan extending effects have been shown in worms, yeast, fruit flies, and mice. A later study in rats was able to demonstrate an amazing 44% increase in lifespan (See the curve below)
It should be pointed out that, although we cannot make our own methionine, the bacteria in our guts can! A neat study in c-elegans worms suggests that the diabetes drug metformin may actually be exerting its lifespan-extending effect by inhibiting methionine metabolism in the gut bacteria of the worm, starving the worm of methionine! This is an interesting paper, as the mechanism of action of metformin has still yet to be agreed upon by the scientific community.
Aging Clocks & DNA Methylation
The current revolution in aging biology is undoubtedly the discovery of the “Aging clock” by Steve Horvath. In a landmark 2011 study, Horvath discovered that you can accurately predict someones chronological age (within about 5 years!) by measuring a handful of methylation markers in the genome. Subsequent to this study, several other papers have developed better models that are even more accurate in predicting age and mortality. These developments have been coined by Horvath as the Epigenetic Clock Theory of Aging.
Epigenetics : This is Your Genome on Meth!
For those unfamiliar, a brief summary of “epigenetics” is the idea that the genome can be modified with markers that don’t affect the underlying sequence. These markers can regulate how DNA is unwound and whether or not specific proteins can bind to it, thereby affecting what genes are activated. As you can see in the wonderful animation by Cell Signaling, there are several such markers/reactions that are used to accomplish this. The aging clocks we are talking about focus on the “methylation” markers that are directly added to the DNA.
Aging clocks work by measuring what sites in the genome have these methylation markers. There are an estimated 28 million sites in the genome that can have this modification, so the landscape is quite complex! Some of these sites are correlated with age, some are not. Some sites show increased methylation with age, and some decreased, but overall, computer models can be used to find the important ones that are the most predictive. Many groups have now demonstrated that a humans age can be guessed, to an accuracy of just a few years, by looking at a handful of these markers. Like counting tree rings!
The epigenetic theory of aging is a powerful new model to understand the potential “cause” of aging. It is generally accepted that epigenetic patterning in the genome help to determine a cells “identity” in the body. In fact, by changing these methylation patterns, you can reprogram cells to act like a different cell type. With regards to aging, its possible that cells are “forgetting”, over time, how to properly behave in their particular local tissue environment.
For an excellent review of Epigenetics and Aging clocks, check out the Sheekey Science show!
Reminder : DNA isn’t the only nucleic acid in cells ….
Plenty of non-biologists read my blog, so a 1 paragraph reminder of the “central dogma of molecular biology” is in order. Our genes store the blueprints for making the protein machines in our cells. Our genes are kept locked up in the cell nucleus (as DNA) for safety, but when the cell needs to make a protein, a “copy” of that gene sequence is made with a slightly different molecule called messenger-RNA. That “mRNA” leaves the nucleus into the cell cytosol where it gets “translated” into a protein. All of this stuff must be tightly controlled and regulated, at every step! Methylation of DNA is one method of control, as we’ve now discussed. RNA must also be tightly regulated in the cell.
RNA is methylated too!
While DNA is hanging out in the cell nucleus, it can get methylated to control which genes get turned “on” in a particular cell. RNA molecules can also become methylated, and these markers have a regulatory role that controls how those mRNA transcripts are translated into proteins.
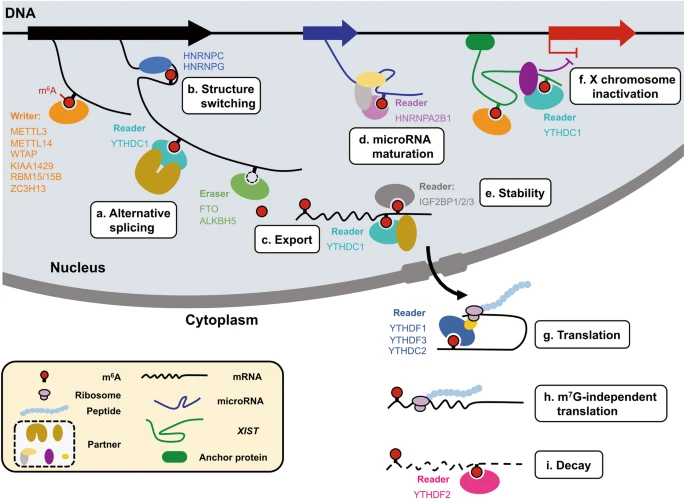
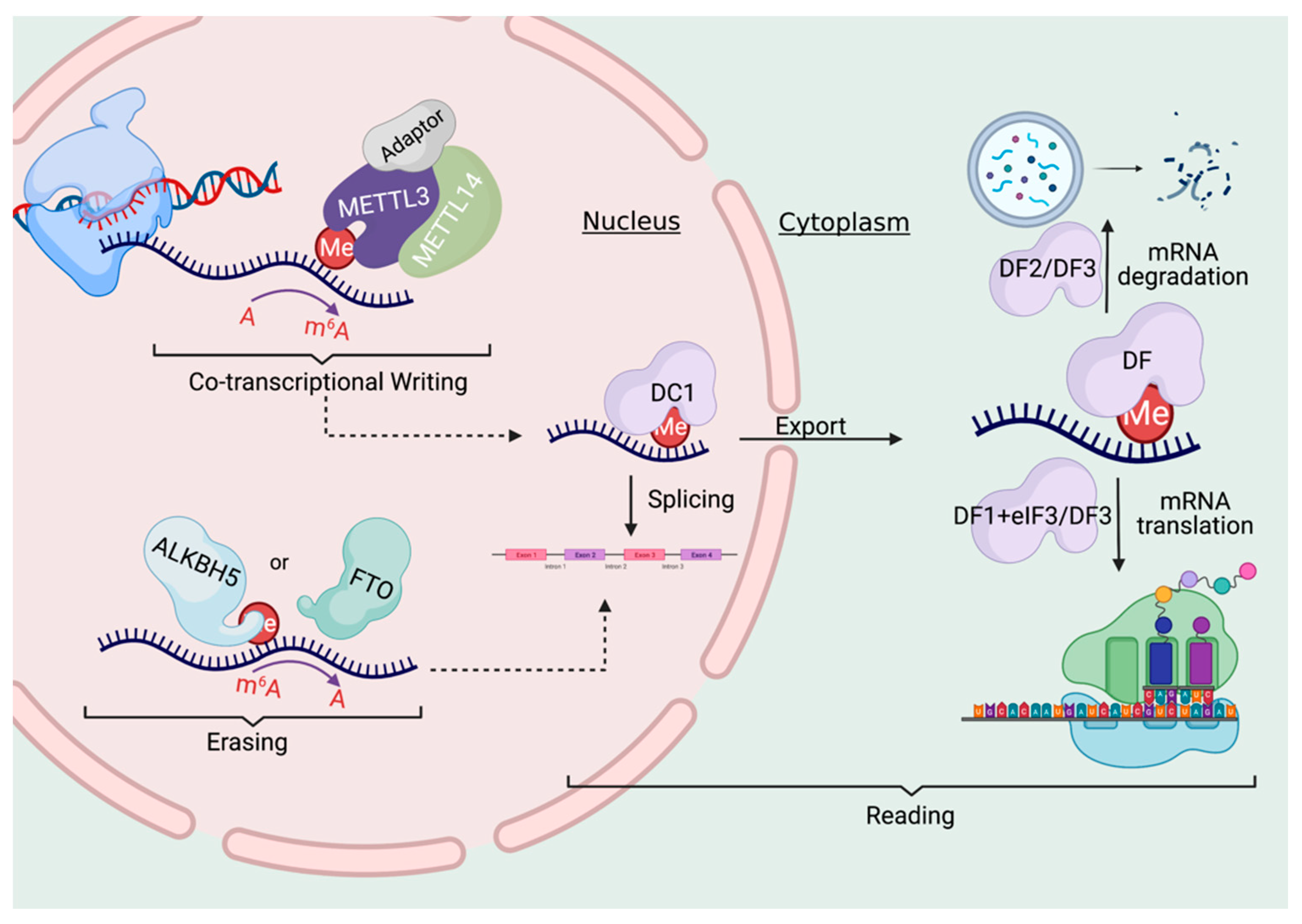
It has been known for decades that a large proportion of RNAs in the cell can be methylated, and the most common modification is the “m6A” methylated adenosine. This m6A marker is added by a large “writer complex” that is formed in the nucleus, right at the site where the mRNA is transcribed from the genome. (Remember this detail for later!) These RNA methylation markers can also be dynamically removed by a set of “eraser” enzymes and also recognized with a family of “reader” proteins.
So what do these methylations do? All kinds of things! They affect export from the nucleus into the cytosol, regulate translation, and mRNA stability/recycling by the cell. That is, RNA methylation, in general, is known to affect how long the RNA sticks around and how much protein is made with its code. If you are an engineer, you should probably be thinking of feedback right about now….
If DNA methylation is associated with aging, is it possible RNA methylation is too? That’s unclear, but I will demonstrate to you that both Obesity and Diabetes, both dominant diseases in the “metabolic syndrome”, have direct links to RNA methylation biology.
So Where Do All These “Methyl” Groups Come From Anyways?
The “methylation” is just the addition of a methyl group. Just a carbon atom and 3 hydrogens. Simple! Methylation markers are abundant in the genome (DNA) and transcriptome (RNA). Those methyl groups have to come from somewhere right? Where?
DNA and RNA are both methylated by a set of methytransferase enzymes. All of these enzyme reactions are dependent on a methyl-donor molecule called S-Adenosyl-Methionine, typically denoted as either SAM, SAMe, or AdoMet. This universal methyl donor molecule is created from ATP and Methionine by the enzyme Methionine Adenosyltransferase (MAT).
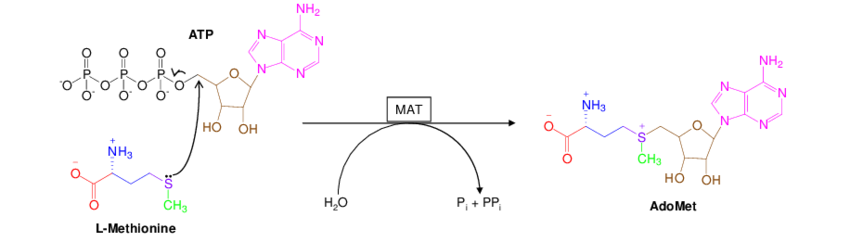
Once the SAM molecule is synthesized in the cell, it is used as the “universal methyl donor” for all the enzymes that need to methylate something. This goes for the DNA cyotsine methyltransferase (DNMT3A), the RNA m6A methyltransferase (METTL3), as well as enymes that methylate histones (another important mechanism of epigenetic regulation).
Speculation zone: Recall that methionine is an essential amino acid. Our cells cannot make it from scratch and we must obtain it from our diets. It’s also a particularly special amino acid since it is the first amino acid in every single protein that our cells make. Given how special and important this molecule is, it would make sense if our cells had a “methionine sensor” to halt protein translation if methionine is scarce. It would also make sense that, if our cells were suddenly starved for energy (ATP), we should stop making proteins immediately. Since Methionine and ATP are needed to make SAM, it’s possible that SAM evolved as a key sensor to centrally regulate protein production (and the DNA/RNA transcription/translation that precedes protein production).
The mTOR connection
On Easter island, the molecule rapamycin was discovered as a fungal metabolite that inhibits the growth of bacteria. This molecule has become an important drug in modern transplant medicine for its immune system modulating effects. Intriguingly, as many of my readers likely are aware, rapamycin is also one of the first drugs discovered to extend the lifespan of several model organisms.
It is thought that mTOR functions as an amino acid sensor in the cell. When it is inhibited by drugs like rapamycin, the cell is effectively fooled into thinking it is starving. This somehow has lifespan-promoting effects that some investigators have suggested is mimicking caloric restriction.
Excitingly, mTOR has recently been shown to directly regulate the synthesis of S-Adenosyl-Methionine (SAM), the universal methyl donor. It does this by tightly regulating the expression of the MAT2A enyzyme that synthesizes it from methionine and ATP. In doing so, mTOR directly controls cellular SAM levels, and thus controls RNA m6A levels and protein translation. Could this mechanism be suggesting that rapamycin’s effect on longevity is mediated via RNA methylation? Possibly!
So here’s where we’ve come thusfar…
Methionine restriction extends lifespan. Methionine is used to manufacture the SAM molecule, which is ultimately the source for the methylation markers added to the genome. The SAM molecule has some spooky links to one of the most famous longevity drug interventions. The recent discovery of “Aging Clocks” have shown that methylation patterns in the genome are predictive for chronological age. Are these interesting connections, or the rantings of a lunatic blogger?
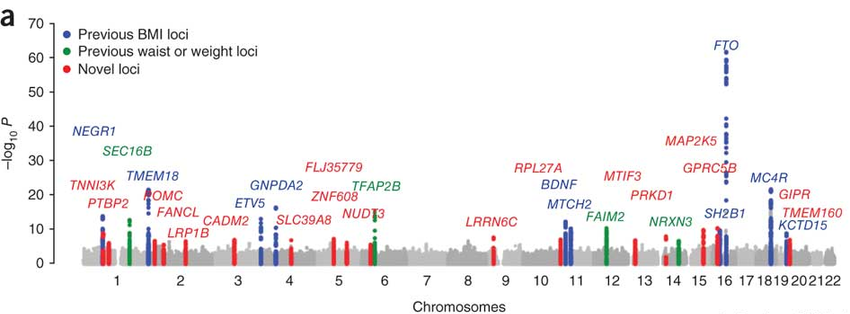
The Genetics of Obesity are tied to RNA Methylation
The manhattan plot below shows the genetic architecture of obesity. Several of the genes below (MC4R, POMC, and BDNF) have all been localized to a specific brain circuit (the arcuate nucleus of the hypothalamus) that regulates food intake and whole body metabolism. That story is an amazing triumph of science that I will write up some day, but it’s too much to discuss in this episode!
For now, we’re going to focus on the single most significant obesity gene locus, FTO.
FTO is an RNA m6a “Eraser”
As previously discussed, N6-methyladenosine (m6A) is the most abundant RNA modification known in cells. When genes are transcribed into RNA copies, they are immediately methylated with an m6a “writer complex” that forms right at the site of transcription. This complex contains enzymes that transfer methyl groups from available S-adenosyl-methionine (SAM) directly onto the mRNA transcripts.
That FTO gene, the top genetic risk of obesity, has been found to be an mRNA methylation “eraser”. It hangs out in the nucleus and specifically removes m6A methylation markers from RNA strands. Whatever it is that RNA methylation is doing with respect to body mass, the FTO eraser counter-acts those modifications. Since its the dominant genetic risk factor for obesity, scientists know that it must be doing something critically important in regulating body mass. We just don’t yet understand the mechanism!
FTO Also Erases the Special m6Am mRNA “Cap”
RNA “m6a” modifications are the most abundant known changes to mRNA. They are found throughout the length of mRNA strands, but it is now known that some may be more important than others. Recent work from the Jaffrey lab in the last few years has found that a specific m6A site is of critical importance! This lab has shown that many mRNA transcripts start with a special m6A adenosine that has an extremely important role in the regulation of transcripts.
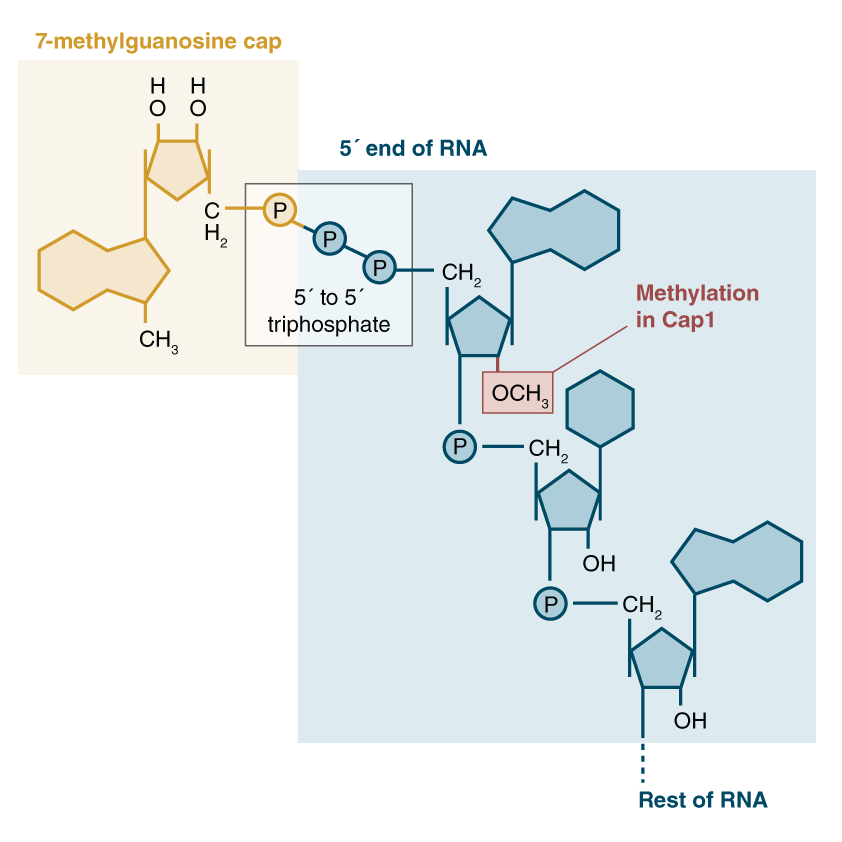
During transcription, both ends of mRNA strands are modified to facilitate their export and trafficking out of the cells nucleus. The “front” (5′) end of the mRNA strand has a “cap” molecule added to it consisting of a 7-methyl-guanosine nucleotide (7mg) with a bridge of 3 phosphates. After this bridge, the first nucleotides of the mRNA are typically modified as well. In many cases, the first nucleotide is commonly a methylated m6a adenosine, that is then methylated a second time on the ribose sugar. This special doubly-methylated m6a at the start of the mRNA sequence is known as an m6Am. The first “m” (m6Am) refers to the methyl group added to the adenosine ring (as in other m6A nucleotides), while the second “m” (m6Am) refers to the ribose methylation added to the sugar backbone. Thus, m6Am.
The 6-methylguanosine “cap” has a critical function of protecting the mRNA strand from digestion by cellular ribonucleases, which would otherwise immediately start eating the mRNA from the 5′ end like a voraciously hungry piranha 🐟. Our cells are loaded with these RNA cleanup enzymes.
The cap isn’t permanent however. The cell eventually will need to completely degrade mRNA transcripts once it no longer needs to make that particular protein. To do this, the cell relies on the DCP2 enzyme to perform RNA de-capping. Once complete, the mRNA is readily destroyed by the ribonuclease piranhas 🐟.
While the 7mg cap’s mission is to protect the mRNA strand, it appears that the very first RNA nucleotide has a regulatory role in protecting the cap! Jaffrey et al showed that the first nucleotide, often a methylated m6Am, prevents the de-capping enzyme from binding to the strand! He further went on to demonstrate that the dominant obesity gene, FTO, preferentially removes the methylation from that specific m6Am nucleotide! That is, the FTO gene is effectively a kill switch that marks RNA transcripts for destruction when they are no longer needed.
The mRNA cap has another function relevant to this discussion. It directly targets the RNA to the translation machinery of the cell. When the 7mg cap is present, it gets bound by a series of cap-binding complexes. One such complex binds in the nucleus and helps to escort the mRNA out into the cytosol. Once in the cytosol, the eukaryotic initiation factor 4 (eIF4) complex instead binds to the mRNA cap, and this complex in turn binds to a ribosome pre-initiation complex that is standing by, waiting to make a protein! (That pre-initiation complex comes pre-loaded with a methionine-tRNA, ready to set down the foundation for a new protein!)
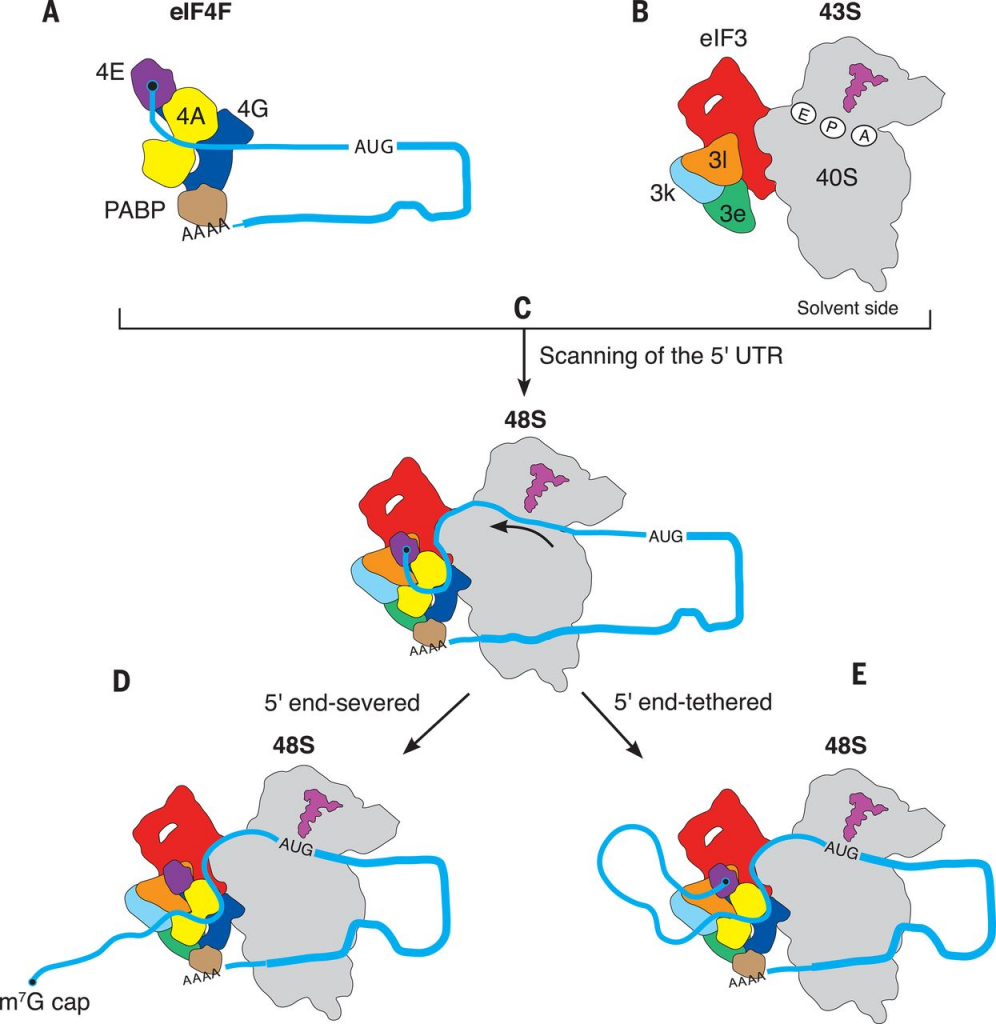
So effectively, the 7-methyguanosine mRNA cap is rather critical for making proteins, and binds to cap binding complexes to facilitate these motions. When present, the mRNA gets trafficked to the ribosomes. The other key purpose of the 7mg cap is to protect the mRNA from degradation. Without that cap present, the mRNA would be rapidly attacked by mRNA nucleases that chew on the 5′ end of the RNA. When the cell is done with an mRNA transcript, it “de-caps” it with the enzyme DCP2 so that it can be degraded by these enzymes.
RNA caps as translation “on switches”
This whole pathway is a bit roundabout, but here is my attempt to interpret what is going on. The RNA cap, defined by the special 7mg nucleotide, is akin to a molecular on-switch for that particular gene transcript. That cap functions by 1) blocking degradation by nucleases and 2) specifically binding the mRNA strand to the translation machinery. Removing that “on switch” with the mRNA de-capping mechanism is akin to disabling the need to make more of that gene into protein.
The m6Am nucleotide, often immediately flanking the 7mg cap, apparently resists decapping. The FTO gene, key to obesity, removes the N6-methyl group from that nucleotide, thus allowing decapping to proceed, which in turn immediately halts translation of that gene transcript.
Thinking of methionine again, as the origin of those methyl groups during transcription, is it possible that FTO here is acting as a cellular starvation sensor that immediately halts translation for particular genes?
Evidence that FTO is a “Nutrient Sensor”
Given that FTO is the most important gene in obesity, it would make obvious sense that it has something to do with sensing of available nutrients. Many in the literature have explored this possibility. Most interesting to me are the specific papers exploring its role in sensing protein.
In one of the earliest characterizations of FTO’s enzymatic activity, it was shown to be expressed throughout the body, but particularly abundant in the arcuate nucleus of the hypothalamus. The authors thus focused their attention there, since this is the same location where several other obesity-related genes (MC4R, POMC, BDNF) have been characterized. They determined that the enzyme is dependent on the cofactor 2-oxoglutarate, also known as alpha-ketoglutarate, a rate-limiting metabolite in the krebs cycle, and thus potentially an important sensor of mitochondrial metabolism. They also demonstrated that the expression levels of this gene vary widely with feeding status. FTO expression dropped 60% during a 48 hour fast in mice! This hints that FTO could potentially be a sensor of starvation, and its role in that key hypothalamic nucleus could be central to regulating whole body metabolism and appetite.
This paper also discusses a key experiment involving leptin signaling. Since the arcuate nucleus of the hypothalamus is known to be responsible for sensing circulating leptin in the blood, a hormone produced by fat cells, the authors hypothesized that injecting leptin into starved mice might restore the levels of FTO expression. It did not, suggesting that FTO is not sensing stored body fat, but something else.
Years later, the same scientists showed that FTO expression was influenced by the availability of amino acids in cells. In a second paper, they showed that FTO was specifically down-regulated by the deprivation of essential amino acids, as opposed to non-essential amino acids. They further tested a few specific combinations of amino acids, and found that depriving Methionine & Cysteine, together, produced a substantial drop in FTO expression.
A previous theory, that I am now second guessing!
Four years ago I wrote about fascinating evidence that has accumulated that some viruses can cause obesity in animals by reprogramming fat cells. In that article, I also briefly touched upon the emerging field of study known as viral epitranscriptomics, which hypothesizes that RNA methylation evolved as a cellular antivirus defense. I think there still could be some overlapping explanations here, given that viruses may merely be hijacking cellular nutrient sensing in order to boost their replication.
I previously discussed RNA ribonucleases as the hungry cellular piranhas 🐟 that eat RNA molecules end-first with no delay! These ribonucleases are a key part of our cellular defense against RNA viruses, whose payloads we do not want to convert to proteins. The 7mg cap is thought by many to have potentially evolved for this purpose. By relying on this mechanism to better distinguish cellular from viral mRNA, it is still possible that FTO plays a role in viral-induced obesity.
The Genetics of Type 2 Diabetes & RNA Methylation
Obesity isn’t the only feature of metabolic syndrome. What about diabetes?!
In a previous blog post, I discussed the genetic architecture of Type 2 Diabetes risk, and primarily focused on the top locus, TCF7L2, a fascinating transcription factor that regulates insulin signaling pathways. Its a fascinating story and I invite you to check it out, but there are many more loci in this Manhattan plot worth discussing.
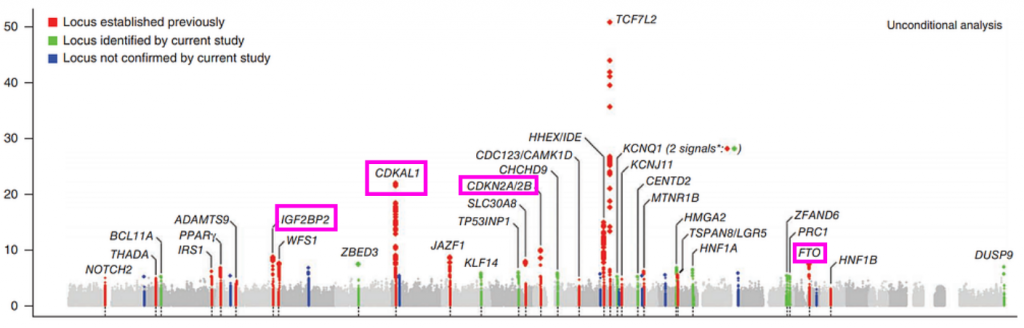
Besides the towering TCF7L2 locus, several other can be seen. We can also see the obesity-associated FTO locus. Furthermore we also see the CDK2N2A/2B locus, which is the key mediator of cellular senescence and additionally the most associated locus with heart disease. These loci make sense given that obesity, diabetes, and heart disease are all comorbidities of the “metabolic syndrome” and tend to influence eachother. The CDKN2A locus also ties type 2 diabetes to what we know about cellular aging and senescence.
CDKAL1 and IGFBP2 Genes are tied to RNA Methylation
The CDKAL1 locus is the second highest peak in this manhattan plot “skyline”. This is a fascinating enzyme that I will eventually need to devote an entire page to. The short story is that CDKAL1 is an RNA methyl-thiol transferase! It doesn’t just add a methyl, but a methyl plus a little extra sulfur group as well! Instead of modifying messenger RNAs (mRNAs) like FTO does, CDKAL1 modifies transfer RNAs (tRNAs) that are needed for protein translation!
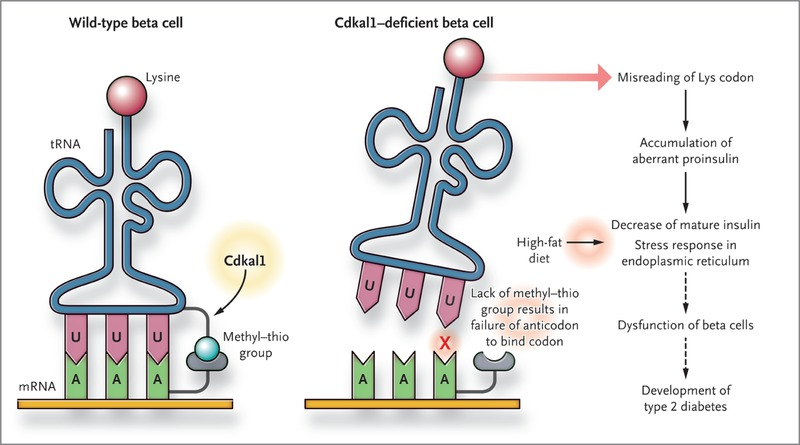
Its not entirely obvious how this gene would relate to diabetes in particular, but the best evidence found thusfar is that the proper functioning of this gene is needed to make functional insulin in the pancreas. When this pathway breaks, the insulin protein is translated with errors that make it impossible to process into its active form. I’ll save the details for a future writeup.
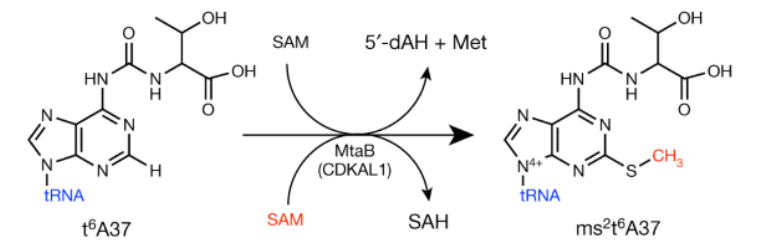
Relevant to this discussion however, here we have yet another enzyme that adds a methyl group (plus an extra thiol) to a nucleic acid. Where does it get these parts? CDKAL1 is dependent on not one, but two S-adenosyl-methionine (SAM) donors to make this reaction work!
The last locus I want to discuss is the IGF2BP2 gene. This locus is nowhere near as strong of a signal as TCF7L2 or CDKAL1 genes, but it turns out to be yet another m6a “reader” protein. That is, it binds to the same m6a methylation markers in mRNA transcripts that the FTO obesity gene removes. Most articles seem to suggest that the dominant role of the IGF2BP genes (1, 2, and 3) is to stabilize mRNA transcripts, which prevents their degradation.
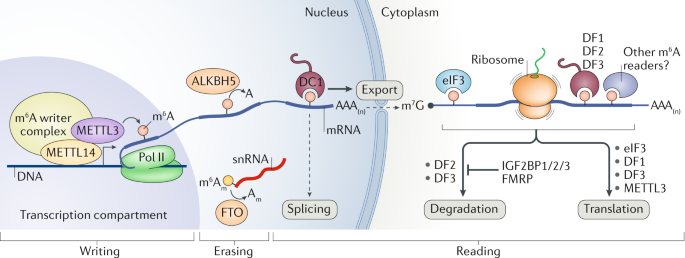
Speculation zone : If IGF2BP2 binds to mRNAs that are decorated with m6a methylation markers, this would seem to suggest that the more methylation on an mRNA transcript, the longer it “sticks around” to be made into proteins. Does this make sense? I would think so! If you have lots of methionine around you can make more proteins. If you are methionine starved, it might be time to recycle transcripts since the ribosomes won’t be able to translate them anyways. How does this relate to Type 2 Diabetes? I can only guess! From my time during my PhD studies of insulin signaling, it was really driven home to me that the insulin signaling axis controls both glucose uptake AND protein synthesis. When the body is starved of glucose, muscles become insulin resistant and break down their proteins so that amino acids can be sent to the liver, whereby they are converted into glucose in a process called gluconeogenesis. Glucose metabolism and protein synthesis/degradation are linked.
Similarly, in a methionine-deficient state, perhaps the CDKAL1 gene evolved to sense this state and produces proinsulin that cannot be properly processed. Thereby reducing the insulin secretion and preparing the body to increase peripheral proteolysis and subsequent hepatic gluconeogenesis.
Intermission
So far we’ve discussed that our genomes, composed of DNA, can be decorated with methylation marks that control which genes are turned on and off. Amazingly, the pattern of these markers forms a sort of “code”. “Aging Clocks” are models fit to these DNA methylation patterns. Using them, you can predict someones chronological age, like counting tree rings! This discovery has ushered in a new “Epigenetic Theory of Aging”, and hinted at future therapies for reprogramming cells into a more youthful state.
RNA is also methylated in several different ways. While DNA methylation has been shown to regulate how genes are transcribed, RNA methylation regulates how those gene transcripts are translated to proteins. Obesity and diabetes, two “diseases of aging”, have deep genetic links to RNA methylation pathways.
It would be cool if we could connect metabolic syndrome, and its associated diseases of old age, with “aging” itself. Caloric restriction, and then later methionine restriction, have left us important clues here that hacking metabolism can extend lifespan. Similarly, drug interventions like rapamycin, that affect protein sensing via mTOR, can also extend lifespan! Given that we cannot synthesize methionine, and that methionine is the ultimate source of the methylations in our DNA and RNA, could this hint that all of these pathways are linked?
Wouldn’t it be neat if DNA methylation and RNA methylation were linked? If aging clocks are correct, then the “methionine code” within our DNA must have some way of knowing how much time has passed. Cells have been shown to have internal circadian clocks, that can track time over a day, but extending this timekeeping to a human lifetime is quite a feat! The DNA molecule, all by itself, cannot keep time. The methylation patterns on DNA must be “written” by the cell in a manner that reflects the passage of time. Finding these external processes, and their link to either age, or a process of “aging” is what we must decipher. Wouldn’t it be neat if these two “methionine codes” could pass information between each other?
DNA and RNA methylation are Interlinked!
Just 8 months ago, as I write this, a fascinating paper was published in Nature Genetics that found a mechanistic link between DNA methylation and RNA methylation. The paper is extremely dense, as they had to go to incredible lengths to establish what is a fairly complex process, but the summary is that they found a mechanism by which RNA m6A markers can drive the de-methylation of DNA CpG sites!
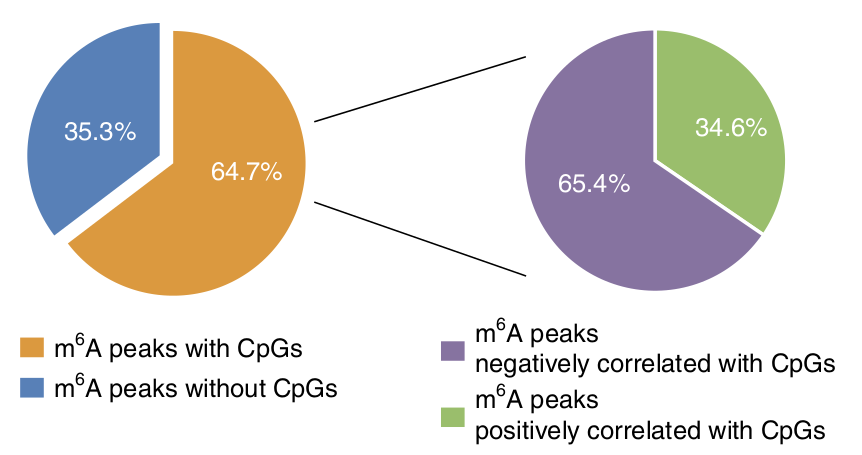
The paper begins with a discussion of a bioinformatics analysis that they performed on existing RNA and DNA methylation datasets. They found that for a large fraction of genetic loci, CpG methylation sites in the DNA tend to occur near (within 2kilobase) regions of m6A methylation in the RNA transcripts, although they also found that the level of these methylations are negatively correlated in 2/3 of all loci. It is important to recognize that this clustering of DNA CpG sites and RNA m6A sites are found on a per-gene basis. If this pattern is real, understanding these gene sets may be critical to better understand which cellular pathways are regulated by this mechanism.
Time to knockdown some genes!
With this bioinformatic analysis suggesting that RNA and DNA methylation may influence eachother, the authors then went about knocking down important genes to see what methylation patterns could be modified. The focused on genes that partake in both the writing and erasing of m6A markers. (See below)
As discussed previously, RNA methylation markers are created by a “writer” complex that forms at the site of DNA transcription. The METTL3 and METTL14 genes are critical components of this complex, so the authors knocked these down to observe the effects. When METTL3 is knocked down, they found that the vast majority of RNA methylation sites decreased, as can be expected. Intriguingly however, this knockdown of an RNA methyltransferase also changed DNA methylation levels, with a large proportion of DNA methylation sites increasing.
They also knocked out METTL14, another key component of the writer complex, and assessed global DNA methylation levels in three different cell lines. In each experiment, global DNA methylation increased by about twofold. They then knocked out the FTO gene, which recall is an RNA m6A “eraser” gene and is also the most important genetic factor behind obesity. With FTO knocked out, global DNA methylation levels fell! That is, this RNA demethylase appears to be regulating the DNA epigenome!
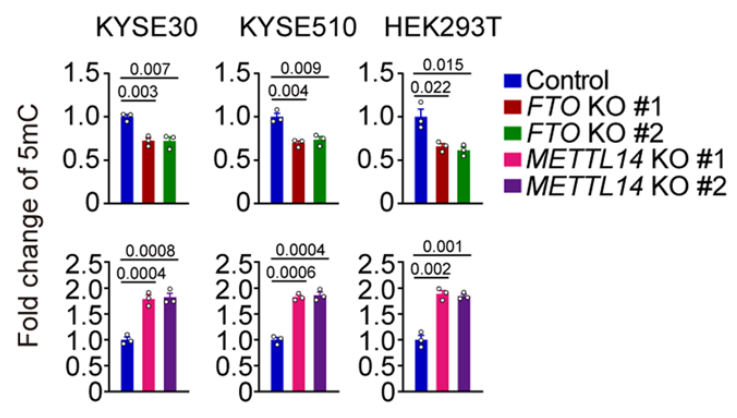
This is truly amazing! These knockdown experiments messed with genes that write or erase RNA methylation markers, yet they had profound effects on DNA methylation patterns! This strongly hints that RNA methylation markers are effectively “talking back” to the genome! Perhaps a feedback mechanism!
Finding the Machinery
The authors set out to discover how this RNA -> DNA communication is taking place, and through a set of thorough experiments, have come up with the following model. When DNA is transcribed into RNA, the m6A methylation markers can be written right at the site of that genes location in the genome.
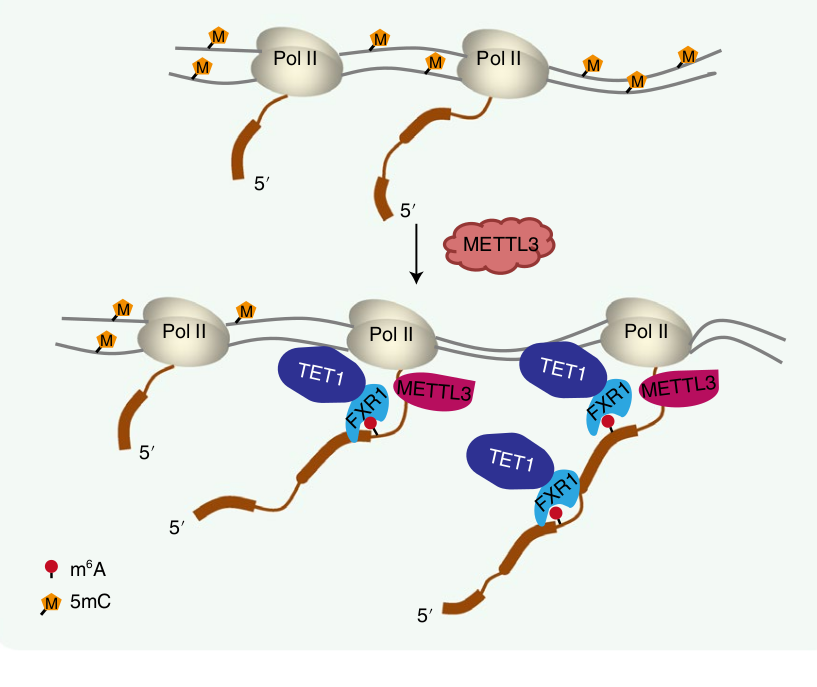
Several proteins are known to bind to these m6A markers, and one in particular is FXR1. That protein, in turn, can bind to the TET1 protein, which just so happens to be the “eraser”of DNA methylation markers!
Importantly, since this is all happening locally at the site of transcription, we have a situation where a gene’s RNA transcript is primarily influencing the local DNA methylation patterns associated with that same gene in the genome. Put another way, the central dogma is running backwards here!
Hypothesis : Nutrient Sensing and Genetic “Feedback”
Speculation zone: It’s generally accepted that cells must precisely control the amount of each protein in the cell, and there is clearly many layers to this. DNA must be unwound correctly, at the right time, and the right locations in the genome for each cell type in the body. Transcript levels must be tightly regulated, for each gene uniquely, and their passage to protein translation must similarly be tightly regulated. All of these dynamics are further affected by nutrient availability, which could potentially change rapidly during times of starvation or stress. In all of these circumstances, each gene must be independently fine-tuned. Global regulatory processes are in play, but also feedback mechanisms at the single-gene level.
So what’s going on here? Why is it that, for a large fraction of genes, high levels of RNA methylation promote DNA demethylation at the site of that genes locus within the DNA? One hypothesis is that each gene in our genome can keep its own internal record-keeping of available cellular methionine, which is the critical first amino acid of every synthesized protein. During times of methionine abundance, higher cellular levels of S-adenosyl-methionine (SAM) could result in higher levels of RNA m6A methylation for a specific gene that is undergoing active transcription. Once formed, m6A sites recruit TET1 to that specific genes’ locus within the genome to demethylate it, making it more likely to be further transcribed. During times of methionine starvation, no recruitment would occur, potentially leaving the methylation levels of that locus unchanged, in the near term.
In short, this is all just speculation, but this RNA->DNA methylation pathway may be a gene-specific feedback mechanism by which nutrient abundance is recorded. The fact that the obesity gene FTO counteracts these m6A markers is intriguing, especially since the authors of the last paper demonstrated that FTO knockout can globally reduce DNA methylation levels. Given the strong evidence we have that methionine restriction can extend an organisms lifespan, it is possible that DNA methylation is being reprogrammed precisely through this nutrient sensing pathway.
The Future – Where do we go from here ?
Promising supplements
While writing this long blog post, it became clear to me that this topic needed its own unified section. There are two key supplements that have been investigated for their longevity-promoting effects, and both directly tie to these pathways Ive discussed.
Alpha Ketoglutarate (AKG)
In recent years, awesome work by Gordon Lithgow, Brian Kennedy, and many others, has shown that this molecule has lifespan-extending properties in model organisms. The average lifespan increase in mice was a modest %12, but they noticed a more prominent effect on overall health. They argue that, since AKG decreases with age, it may be a promising dietary intervention.
Watch this excellent review from Sheekey Science about these particular longevity experiments.
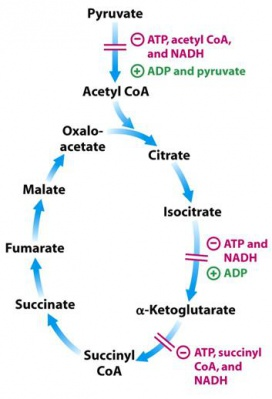
Alpha ketoglutarate is a fascinating metabolite. As mentioned previously, it is a required cofactor for the FTO obesity gene. It is also the required cofactor for the DNA demethylase TET1, as well as the cofactor needed to demethylate histones. If you want to demethylate DNA, RNA, or histone proteins, the cell needs alpha ketoglutarate!
Alpha ketoglutarate is one of the key steps in the krebs cycle, the set of key reactions in our mitochondria that produces most of our cellular energy. Most interestingly however, AKG is right in the center of two key control points of the TCA cycle. It is formed by Isocitrate dehydrogenase, an enzyme that is inhibited by high ATP concentrations. Immediately after AKG in the Krebs cycle is the Alpha Ketoglutarate Dehydrogenase complex, which converts AKG to Succinyl-COA. It too is inhibited by ATP. Because the AKG metabolite finds itself in this key regulatory node in the cycle, it is potentially a sensor for metabolic state by the enzymes that rely on it as a cofactor. Additional studies point to evidence that it is a sensor of redox stress.
The obesity connections to AKG, and the longevity benefits of AKG in model organisms, is a hint that this pathway must be further scrutinized.
S-Adenosyl-Homocysteine (SAH)
An intriguing paper shows that the metabolite S-Adenosyl-Homocysteine has lifespan-extending effects in both yeast and c-elegans roundworms. The paper presents the central thesis that these effects are mediated through the same pathways by which methionine restriction works. They point to evidence suggesting AMPK activation as a key cause of the effects.
S-adenosyl-homocysteine is interesting to me because it is the byproduct of the methylation reactions that modify both DNA and RNA. It is effectively formed when the “methyl” is removed from S-adenosyl-methionine. SAH is what remains.
When SAH levels are high, they can actually inhibit the methyltransferase enzymes that produce it. It has been specifically shown to inhibit the METTL3 enzyme that is the core of the m6A writer complex that methylates RNA. Unsurprisingly, SAH also inhibits the DNA methyltransferase DNMT1, which writes the CpG sites to the genome. Drugs are being developed that are analogues of SAH for anti-cancer use. Time will tell if pharmacological targeting of this pathway could potentially effect aging as well!
Promising Dietary Interventions
I’ve already discussed methionine restriction here as a potent intervention that is now demonstrated in several animal models. The easiest way to achieve this is to stick with specific plant-based foods. As time goes on, I would not be surprised if low-methionine recipes become as common as other popular dietary restrictions.
Methionine restricted diets are being experimented with in cancer treatment. Given the optimistic data that Valter Longo has collected showing that fasting can help with cancer survival, I would not be surprised if CR and methionine restriction both become the standard of cancer care. If these strategies are proven effective, low-methionine hospital meals would be commoditized and more accessible.
When the low-fat craze took hold to fight heart disease, we engineered our food in creative ways. Engineering methionine out might be the next big achievement of the food industry.
Desired Pharmacological Targets
Given that FTO is 1) the dominant gene in obesity, and 2) apparently regulates DNA methylation indirectly via the RNA->DNA feedback pathway, it is clearly an important target for future drug development. Time will tell, but if we ever successfully target it to treat obesity, it would be amazing if it also had anti-aging properties as well. Time will tell.
Knocking out FTO completely is embryonically lethal, and over-expressing it has promoted weight gain in lab animals via increased food intake. It is clear that its expression must be precisely controlled for proper development. Despite this, I’m optimistic that it can be potentially targeted with drugs after development, particularly during later life.
Excitingly, FTO inhibitors have recently been developed as cancer therapeutics. The next few years may be exciting if new experiments find metabolic or anti-aging benefits to these compounds. Buckle up.
Experiments Needed!
While diving deep into these pathways has allowed me to connect a few dots I’ve been thinking about for more than a decade, it has also created more questions than it answered. Many experiments are needed to properly develop this theory further.
First and foremost, we need more studies that assess the Aging Clock on organisms that have undergone caloric restriction and methionine restriction. Ive attempted to find clear studies that have shown this, but the results haven’t been clear. The most appropriate way to conduct this study would be to measure the aging clock in multiple organs, especially the hypothalamus where the key nutrient sensing neurons are located. Several have postulated that hypothalamic aging may be the central regulator of organismal aging, and I fall firmly within that camp given my focus on obesity physiology.
Conclusion
Taking this deep dive has given me much excitement for the future of anti-aging therapies. Ive focused my career on obesity for personal reasons, but have always been fascinating by aging. I think some of these pathways point to underlying connections. The advent of DNA clocks has given me a refreshingly optimistic look for the future of the field.

