Cholesterol: Have we Shot the Messenger?
A followup from : Could Aging be an Infectious Disease?
For decades, we have been fixated on cholesterol as a major factor influencing human health and mortality. It is well established that cholesterol (actually, cholesterol-transporting lipoproteins) come in “good” and “bad” forms, as the relative levels of these variants is predictive of heart disease. Cholesterol also seems to take center stage in Alzheimer’s since the APOE gene is a lipoprotein that transports cholesterol in the brain.
All of this evidence has contributed to the lipid hypothesis, which has underlined much of public health policy and dietary dogma, and indeed driven the original war between Hi-Carb and Hi-Fat diets. The accumulated evidence is strong, but what if we are inadvertently blaming our bodies natural gene when something far more malicious could be present?
What if its not the cholesterol, but something else these lipoparticles carry? In our last suspenseful episode, we reviewed decades of literature demonstrating links between several common viral infections and the development of both heart disease and Alzheimer’s. Just a few days ago, even more astounding evidence was published demonstrating that amyloid-beta in the brain serves an important role in stopping the spread of herpes virus from neuron to neuron.
Today, we’ll dive deep into even more startling connections that challenge some of our deepest and long-held dogmas.
Getting into the cell
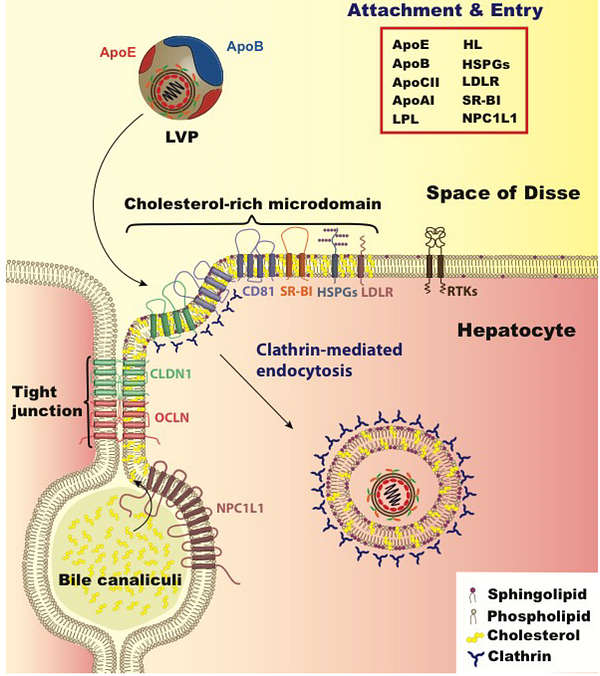
Like a computer virus, a biological virus must get into its host without being detected. What better way than to walk right into the front door with a clever disguise? In computer security, this happens all the time, like an innocent looking email that can break an entire democracy.
In human illness, a profoundly interesting example of this phenomenon is Hepatitis-C virus (HCV). When replicating, it packages itself into a low-density-lipoprotein (LDL) particle, forming a “lipoviralparticle”. These packages then get into cells through the normal endocytic route that is used to deliver cholesterol to all cells of our body, by docking to receptors in a special region of the cell membrane known as a “membrane raft”.
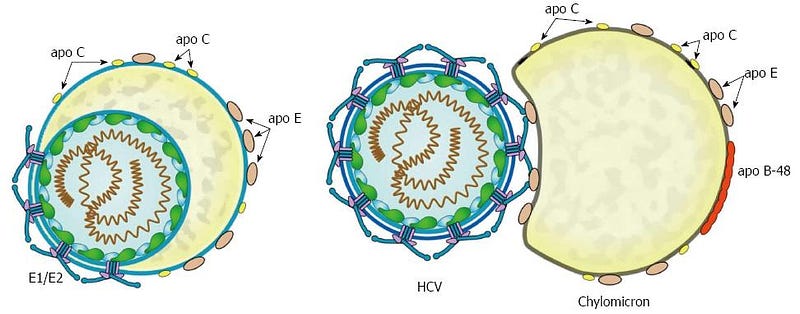
Amazingly, part of the Hepatitis-C spread is reliant on the APOE protein, which is the top genetic association with Alzheimer’s. APOE has been shown to exchange from lipoparticles onto viral-infected lipoviral particles, and when this happens, the lipoviral particle is better able to avoid the immune system. APOE also appears to play an important role in the development of liver cancer after hepatitis-B infection, and the genotype of APOE influences this risk. APOE, in this instance, is serving as a convincing secret agent mustache disguise for both Hep-C and HepB viruses…..
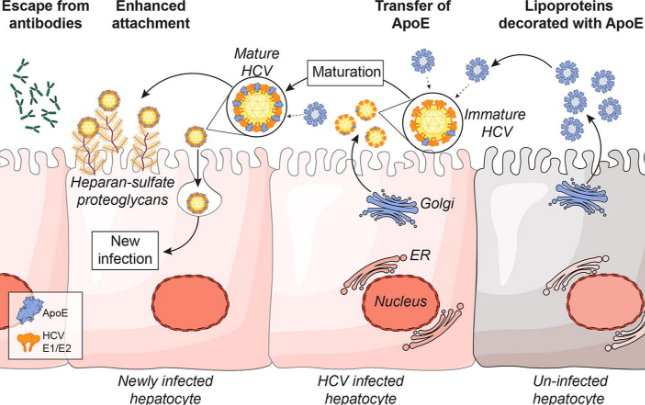
Hepatitis C virus thankfully has a relatively rare incidence of 1 in 100k US persons. Hepatitis B is more common, and is present in 4 out of every 1000 in the US. The human Rhinovirus, however, is the cause of the common cold that we all annoyingly catch from time to time. Totally harmless, right?
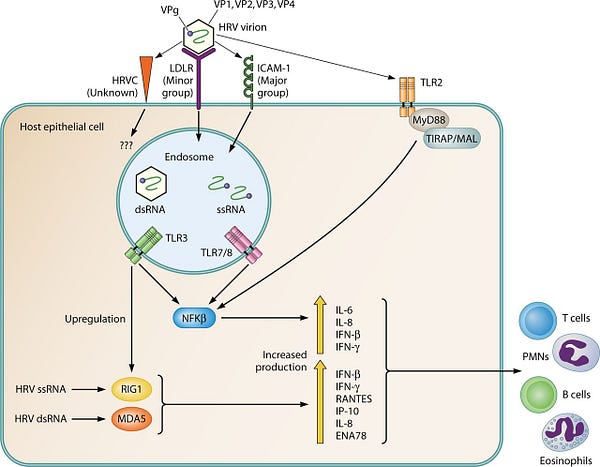
Well, Rhinovirus specifically exploits the very low density lipoprotein receptor (vLDLR), effectively entering the target cells through the exact same front door. Once the viral capsule binds, it exploits the cells endocytic machinery to enter the cell and begin replicating. This endosomal trafficking also occurs at membrane rafts.
While most of us do not take seriously respiratory infections like influenza or the common cold, it should be noted that the risk of having a heart attack increases 14-fold in the weeks surrounding such infections!
Lately however, it is herpes simplex virus that has been getting all the attention as a possible cause of Alzheimer’s disease, and as we discussed last time, herpes viruses have been shown for decades to be causative for atherosclerosis. It has also been demonstrated that herpes viruses bind to several lipoproteins with high affinity, and that they specifically exploit oxidized LDL particles to infect endothelial cells of blood vessels. In the brain, Herpes-Simplex-Virus-1 has been shown to bind to APOE, and it is APOE that allows the trafficking of lipoproteins into cells.
Getting out of the cell
Much like computer viruses, biological viruses must take a delicate approach when they exploit a host. They must avoid detection by the host in order to prevent an Antivirus mechanism from squashing it. They must also avoid killing the host entirely, or they will not proliferate. The most successful viruses, either digital or biological, are those that avoid detection, lurk in their compromised host, and spread as efficiently as possible.
As discussed last time, biological viruses evade the complement system and optimize the cellular metabolic machinery in order to increase their rate of spread. Furthermore, the best biological viruses have evolved to exit our cells as efficiently as possible without killing them.
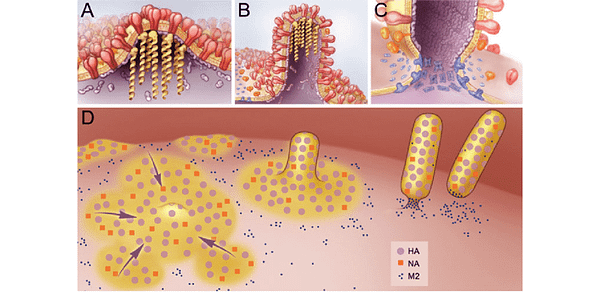
To accomplish this, many viruses have evolved the ability to “bud” from our cell membranes, primarily at the site of membrane rafts, where a high cholesterol content permits the membrane to be highly flexible. For a detailed overview of these mechanisms shared by a great many viral strains, these two papers are excellent resources.
Given that several viruses appear to enter the cell at membrane rafts, exit the cell by “budding” from membrane rafts, and exploit binding to both lipoproteins and lipoprotein receptors to easily get around the body and evade the immune system, is it any wonder that the levels of these lipoproteins may influence health and disease? Indeed, some studies have shown that cholesterol lowering drugs can inhibit the spread of some viruses.
Alzheimer’s connections
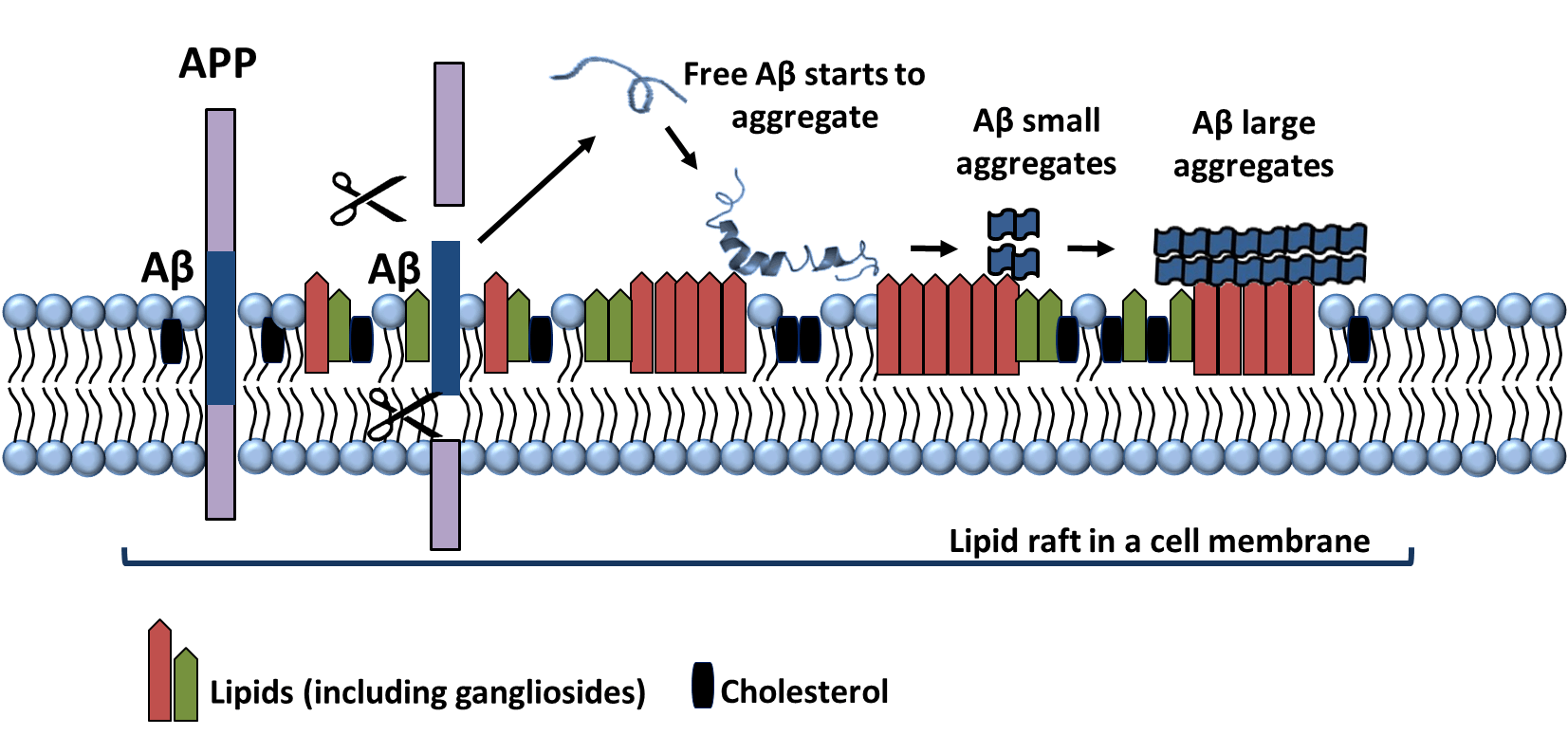
Alzheimer’s is a disease that is associated with the accumulation of two specific proteins: Beta Amyloid and Phosphorylated Tau. With the recent exciting publication demonstrating that Beta-Amyloid functions as a trap for Herpes viral particles, it is worth discussing where the beta-amyloid comes from. Beta Amyloid protein is secreted primarily at the site of membrane rafts, and as the proteins form aggregates, they maintain a tight binding to these rafts via interactions with the specialized lipids that they are composed of, primarily gangliosides.
While the accumulation of beta-amyloid between neurons was the first hallmark that Alois Alzheimer originally noticed, the accumulation of a phosphorylated protein called Tau has received much attention due to its formation of “Neurofibrillary tangles” within the neurons. The primary function of Tau, however, is to regulate the stability of microtubules within cells, which are used to transport cargo around, among many other functions.
Microtubules in neurons are critical for the transport of things along the long-lengths of axons, and motor proteins allow the movement of things in both directions. Anterograde transport moves things towards the synapse at the end of an axon, while retrograde transport moves things back to the cell body. In neurodegenerative disorders, a common theme appears to be defects in this trafficking system, possibly mediated when Tau can no longer do its job.
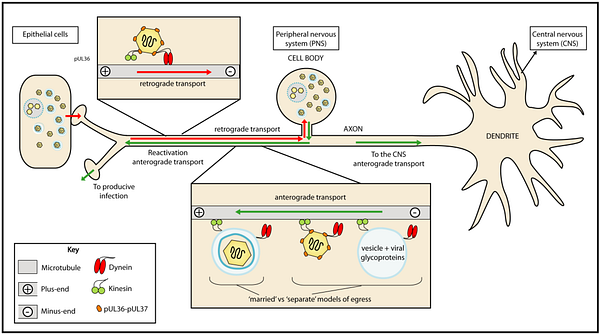
Retrograde signaling, it turns out, is also the primary mechanism by which Herpes and Rabies viruses infect the nervous system. They specifically travel backwards through neural circuits, which many in the literature have proposed as an explanation for how they could enter the brain from the periphery. This transport is actively facilitated by motor proteins that transport virus particles along microtubules.
And this is the part where I propose something with zero evidence to back it up: Could Tau hyperphosphorylation be an attempt by the cell to halt the progress of a virus? Alternatively, could Tau hyperphosphorylation be a mechanism viruses use to make axonal transport more efficient? Interestingly, it was demonstrated years ago that herpes infection causes Tau to become hyperphosphorylated. Despite this find, evidence of Tau’s function in regulating axon transport has been mixed, but experiments do not seem to have not focused on the viral hypothesis.
Back to Aging Biology
As heart disease and Alzheimer’s are both considered to be diseases of aging, it is useful to revisit the cellular understanding of what aging is. Aging has many “hallmark” characteristics, but a dominant model focuses on a state that we call cellular replicative senescence in which a cell becomes permanently incapable of dividing. This is widely assumed to be a defense mechanism against cancer, but it has been shown that senescent cells secrete a variety of inflammatory molecules that can make an entire tissue sick. It has also been shown in mouse models that simply removing these aged cells can boost lifespan by up to 35%! Unsurprisingly, finding drugs that specifically kill senescent cells has become a booming new industry. (See Unity Biotechnology)
As we discussed in our last episode (go read it, seriously!), a common marker for cellular senescence is the p14/p16 gene, which is the top genetic association for heart disease and specifically functions to freeze the cell cycle between the G/S transitions. This cell-cycle checkpoint pathway is exploited by many viruses in order to maximize replication and minimize cell death, including HSV-1.
The second common marker for cellular senescence that has been embraced by the aging research community is activity of a “Senescence Associated Beta Galactosidase”. For many years, this was used as a reliable marker for staining senescent cells, but it was finally traced to a specific gene GLB1, which is a lysosomal beta-galactosidase enzyme. Rare mutations in this gene cause GM1-gangliosidosis, a disease in which GM1 gangliosides build up within the brain. GM1, you may recall, is considered the primary distinguishing lipid contained within membrane rafts.
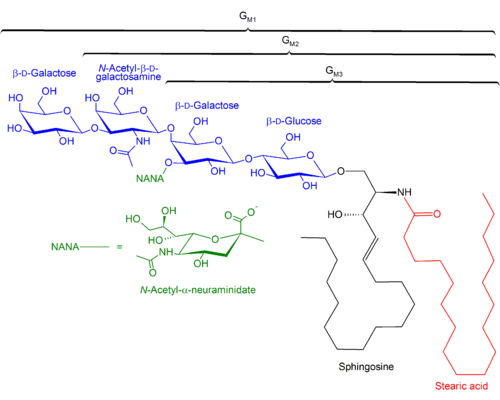
The GM1 lipid headgroup is composed of a galactose sugar, and a special sugar called N-Acetylneuraminic acid, which is also called “Sialic Acid”. Several viruses, including influenza, specifically bind to GM1 molecules at these headgroups. Ironically, influenza viruses exploit an enzyme that degrades neuraminic acid, neuraminidase, to facilitate this binding and allow cell entry. The version of this protein is the “N” in influenza strain designations such as “H5N1”.
Given that in our last writeup, we discussed how p14/p16 expression and cell-cycle locking can be exploited by viral infections, it may be that overexpression of the senescence associated beta-galactosidase (GLB1) is a similarly important antiviral defense. By degrading the population of GM1 gangliosides in a cell, this may potentially limit viral entry in much the same way that beta-amyloid aggregated can prevent export by binding to the external face of the cell.
This second connection between cellular senescence and viral replication warrants followup, and preferably communication between the aging field and virology field.
============ In closing
Have we needlessly blamed our good friend LDL, who has always been there to deliver cholesterol to peripheral tissues in need? Is it possible that we have become far too fixated on simple answers for explaining all of our common health problems, and consequently obsessed over simple solutions (like low-carb or low-fat dieting), when in fact we should focus on more nuanced explanations for what kills us?
Maybe we should stop shooting the messenger, and while we’re at it, stop blaming sick people for their dietary choices?
================

Rorty: I’ve got nothing but bad memories about these lipoproteins Mick. Everytime they show up, it seems that atherosclerosis develops and a heart attack eventually ruins everything.
Mick: That’s because of the parasites Rorty! These viruses are hiding inside our own lipoproteins to sneak around our bodies. They’ve even convinced us to attack our own friends, through autoimmune reactions!
Lets go, we’ve got lots of our own healthy lipoparticles to exterminate.
=================