It’s high time I finally get around to telling you about my favorite protein, one that I originally stumbled upon in my Diabetes PhD at ASU and the Mayo Clinic over 6 years ago. Ever since, I have been slowly deciphering what exactly is going on. I’ll be summarizing some parts of my dissertation, so this might be a long one, but its worth it….
At the Mayo Clinic, we performed a rather lovely experiment where we obtained glucose clamp data on test subjects, and coupled this with gene expression analysis on muscle biopsies before and after exercise. This work resulted in a great deal of interesting hits, including the pathway I’m going to discuss today.

From that study, we were able to obtain a list of genes that change with exercise in a manner that strongly correlated with insulin sensitivity before exercise. Among the positive correlates was the gene ACP5. For whatever reason, insulin sensitive subjects made more ACP5 post exercise than the insulin resistant (diabetic) subjects.
The ACP5 gene is more commonly named TRAP (Tartrate Resistant Acid Phosphatase). It enzymatically removes phosphate groups from proteins, but the immediate question is, could it have something to do with metabolic syndrome? The first profound clue that it does was a study I found that over-expressed this gene in mice, and the result is glaringly obvious. Turn TRAP on, and mice get GINORMOUS, but more importantly, they remain completely insulin sensitive. IE, they get fat, but not diabetic. WTF? Even more interesting, there are some profound sex differences in this effect, particularly with food intake. Sorry ladies.

So what’s going on here? Years ago I began digging for a mechanism of action, and I learned that in bone, TRAP/ACP5 has a well-characterized function. It’s an important molecule that regulates the attachment of cells that create and destroy bone, the osteoblasts and osteoclasts.

Osteoblasts bind to bone and lay down minerals, while osteoclasts (figure above) bind to bone and secrete acids into a controlled space to dissolve the bone matrix. Among the many things secreted into this space is the ACP5/TRAP protein. To keep the acids contained in the pocket, the cells form a “sealing zone” around the perimeter with the αVβ3 integrin receptor, also known as the vitronectin receptor. This receptor binds to, naturally, the protein known as vitronectin in the bone extracellular matrix. It has a few other binding partners however, and one of them is a little gene called SPP1, also known as bone sialoprotein 1, and osteopontin.
Osteopontin is evidently one of the core regulators of osteoclast binding to bone due to its binding to the αVβ3 receptor, but also appears to mediate the detachment process. In 1994, it was shown that attachment was dependent on osteopontin phosphorylation. The TRAP enzyme pops this phosphate off of osteopontin, and when this happens, the osteoclast can release from the bone.
So the ACP5/TRAP gene, which is strongly associated with insulin sensitivity when it’s activated after exercise, dephosphorylates proteins. Osteopontin is one of them. Who cares?

A) Glucose infusion rate during a glucose clamp. B) GDR is the glucose disposal rate in peripheral tissues, and C) HGO is the hepatic glucose output, from gluconeogenesis.
Aside from bone stuff, what else is osteopontin involved with? Well, scientists decided to go create another franken-mouse. This time, they knocked the osteopontin gene out of the genome to see what happened. What they found was spectacular. These mice were completely protected from a high fat diet.
If you feed a normal mouse a high fat diet, they develop insulin resistance and all of the symptoms we associate with diabetes. Remove osteopontin from the animal, and a high fat diet barely produces any change in insulin sensitivity. No diabetes symptoms!
WAT WAT WAT? (that’s a white-adipose-tissue joke)
In summary, turn on TRAP expression in a mouse, and they get super insulin sensitive. TRAP dephosphorylates osteopontin. If you knock osteopontin out of a mouse, they never get insulin resistant. We also know that osteopontin phosphorylation allows it to bind to the αVβ3 receptor in bone, but only if its phosphorylated. TRAP removes this modification. Could this mechanism have something to do with insulin resistance?
Enter the immune system

Turns out, osteoclasts come from the same line of cells as macrophages, those lovely-but-crazy cells in our body that eat things that they really don’t like. You really want these guys on your side. They can eat bacteria and other pathogens, but they can also eat cholesterol particles, leading to blockage of blood vessels. Understanding the appetite of these immune cells is thus critical for understanding the leading cause of death.
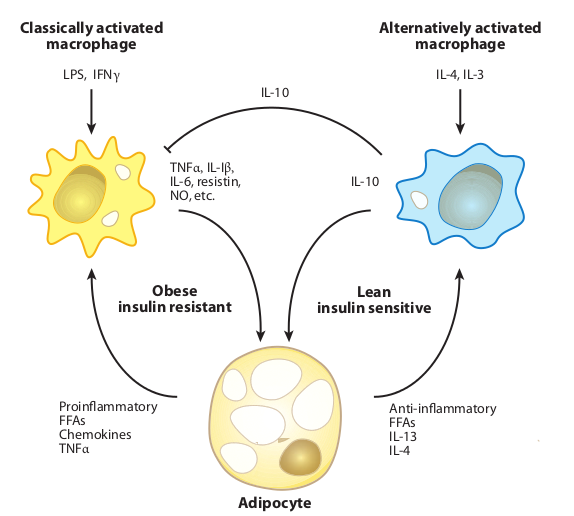
Macrophages can be “activated” into two different states relevant to their police-like activities. There is an M1 (proinflammatory) state that is associated with the secretion of pro-inflammatory cytokines like TNF-alpha, and there is an M2 “calm the fuck down” state associated with wound repair. The M1 state can be easily induced when the macrophages sense bacterial toxins like LPS. I like to think of this M1 state as police arriving on scene with guns drawn to neutralize an invader, whereas the M2 state is the following investigation, medical, and cleanup crews sent in to heal and repair tissues (like after exercise!).
During my PhD this article in particular profoundly changed my outlook on the disease.
Macrophages, and their polarization state, have a direct connection with insulin resistance. Jerrold Olefsky at UCSD has long argued that in fact, macrophages invading muscle, fat, and liver tissue are a central component of diabetes.
As a matter of fact, it has even been directly demonstrated that the amount of macrophages invading into muscle tissue correlates strongly with body mass index, and that insulin sensitivity can be restored in humans when specific drugs are used, which drive away the invading macrophages out of fat tissues.

So, why is it that these macrophages invade these tissues, get polarized into the trigger-happy M1 proinflammatory state, and start shooting? Well, turns out osteopontin is the gene that called in reinforcements. In adipose tissue at least, it directly regulates the inflammatory state of macrophages, and it is generally upregulated in the fat tissue of obese humans.
Even more exciting, it was later shown that it’s specifically osteopontin binding to integrin receptors found in macrophages that regulate their inflammatory state! In this profoundly amazing work, they demonstrated that simply dephosphorlylating osteopontin switches the macrphages into a calmer state, associated with reduced secretion of tumor necrosis alpha (TNF-alpha), which is one of the most potent inducers of insulin resistance in peripheral tissues.
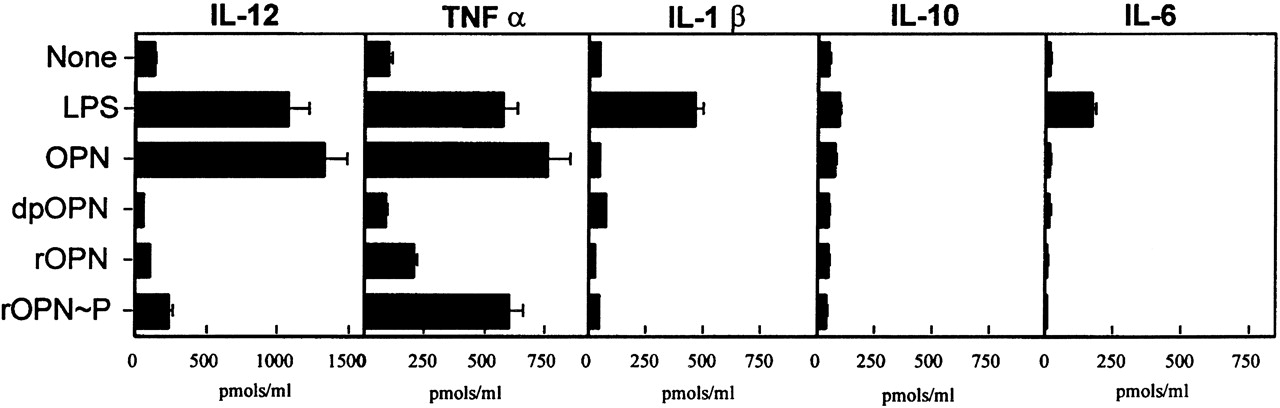
The mechanism at this point appears to be that macrophages are getting recruited into various tissues and triggered into a panic phase. They then secrete proinflammatory cytokines that tell muscle, fat, and liver tissue to become insulin resistant. This happens via the TNF-alpha receptors, and is well characterized. Knock osteopontin out, and mice won’t develop insulin resistance no matter how much fat you feed them. If you instead overexpress TRAP which dephosphorylates osteopontin, mice become completely insulin sensitive. (although fat, because the fat cells are no longer being told to stop taking in glucose…..)
Implications for Exercise
Let’s circle around to the start of this deep-dive. People who are highly insulin sensitive before an exercise intervention, apparently turn this gene on more after exercise than people who are insulin resistant. The mouse overexpression studies seem to indicate that having abundant levels of this gene promotes insulin sensitivity. It is also known that exercise causes a temporary cessation of insulin resistance, which makes sense from the simple perspective of thermodynamics. After all, the muscle needs to replenish the energy it expended.
However, could it be that this core pathway represents an anti-inflammatory benefit to exercise? Could this anti-inflammatory effect be present in some, but not all people?
Future directions
This is the part where I drop the mic with some random factoids about heart disease, which kills 25% of us. I still need to get around to elaborating on these pathways. In doing so, you will be compelled to “stay tuned” for the next post, and I will be compelled to actually write it.
- Osteopontin (SPP1) is highly abundant in coronary artery plaques. In one study, it is the most overexpressed gene!
- Osteopontin appears to have a direct functional role in arterial calcification processes, a dominant feature of heart disease. Makes sense! It was originally discovered to regulate bone turnover!
- The receptor that phosphorylated osteopontin binds to, the αVβ3 vitronectin receptor, has a direct role in macrophages becoming foam cells when they eat LDL particles, which is the primary mechanism causing blockage of arteries. It appears that activation of this receptor switches off the scavenger receptors in the macrophage which allow it to directly target oxidized LDL particles.
You can probably see where this is going. I previously argued that LDL particles can carry viruses around, and this is likely why macrophages eat them in the first place. It should therefore not shock you to learn that it is a key mediator in defending against infections from herpes viruses, which are now implicated in heart disease and Alzheimer’s. With the vitronectin receptor present, herpes viruses can enter cells through the lipid domains associated with cholesterol import.
On the aging biology side, some labs consider oseopontin to be a key member of the Senescence Associated Secretory Phenotype (SASP). I previously argued that cellular senescence might in fact be primarily driven by viral infections, and also discussed how the senescence associated beta galactosidase may in fact be an antiviral defense mechanism because it specifically cleaves sialic acid residues off of GM1 lipids. These sialic acids are important targets for many viruses, especially influenza. Osteopontin is also known as “bone sialoprotein 1” because it has an incredible 10 sialic acid residues on it. Because of this, osteopontin has a rather important role as a decoy for viruses to bind to……