Growing Out of Control
If you are reading this blog, you are likely familiar with the connections between obesity and health conditions such as heart disease, diabetes, and even cancer. You have also undoubtedly heard that obese people generally die sooner than their healthy-weight peers. We all know this, yet the problem continues to grow out of control.
Aside from the health consequences, the obesity epidemic has become one of the most politicized and divisive public health issues of our time. The epidemic has a large societal burden, which has unfortunately been placed upon the victims themselves. The conventional wisdom for decades has followed from the first law of thermodynamics: that people are fat because simply they eat too much and don’t move enough.
Why shouldn’t they carry the burden? It’s their decision right?
NO! It’s really not that simple
Scientific evidence has long-since proven that the body uses a sophisticated set of sensors and control systems to precisely regulate the storage and usage of energy. Obesity results when these control loops break down. This is the consensus among researchers in the field.
These control systems exist at the small scales of individual cells in the body, and as circuits in the most ancient parts of the brain. Most exciting, is that in the last decade of the genomics revolution we have identified a handful of genes that strongly predispose humans for gaining weight, and we are slowly deciphering what they do.
To start our deep dive into obesity, it makes sense to focus first on fat cells themselves. In the lab, we call these “adipocytes”. Like every cell in the body, these cells contain their own control systems that regulate their intake, storage, expenditure, and release of energy. These “decisions” are made by the cell through a set of processes that biologists call “cell signaling”, and involve many different cellular receptors and enzymes that react with eachother. These signaling “pathways” are effectively molecular neural networks, allowing cells to “think”, communicate, and interact with eachother, allowing multicellular organisms like YOU work.
Cellular regulation of energy intake
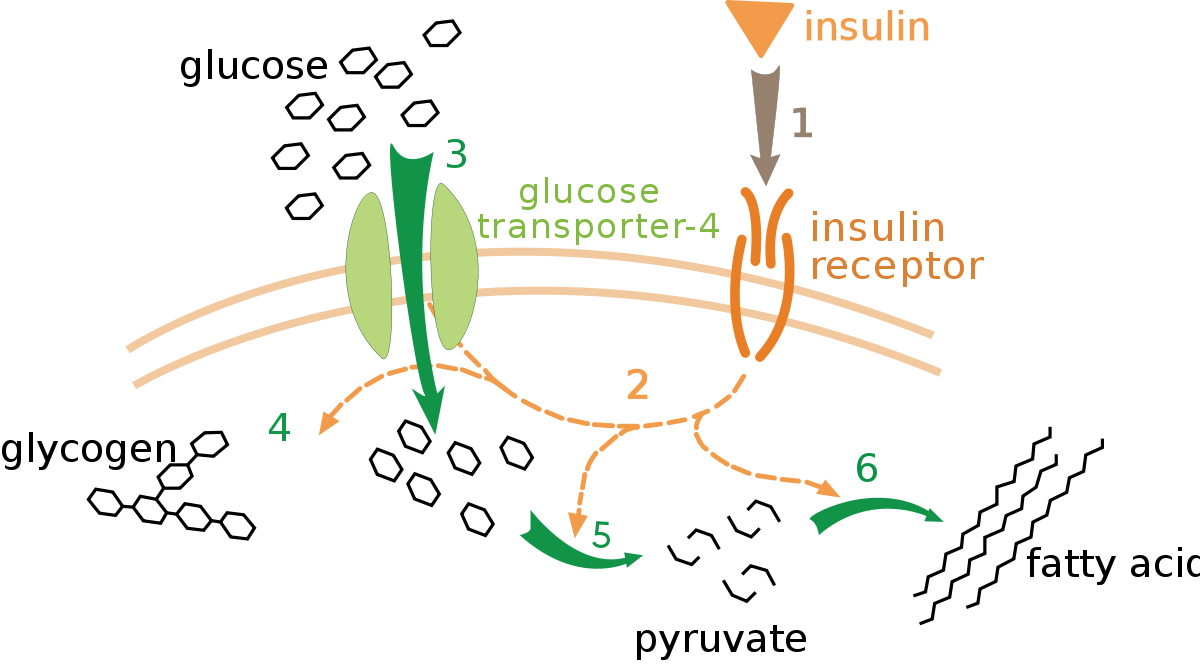
Glucose is a fundamental form of energy in the human body, and its storage and usage is tightly controlled. The body must always keep enough glucose in the blood so that organs like the brain have fuel readily available, but it must also store extra glucose for later if those blood concentrations get too high, since glucose can react with and damage proteins.
This is all regulated via the hormone insulin, which binds to insulin receptors found in cells throughout the body. Insulin binds to a receptor (1) and a “bunch of signaling and thinking” happens (2), followed by glucose transporters getting inserted into the membrane (3) allowing glucose to enter the cell. After this, that glucose can be stored (4) burned (5) or converted to fat (6).
Step (2) in the pathway above is handwavey magic, isn’t it? Somewhere between the insulin receptor and the glucose transporters, “stuff” is happening. After the cell collects insulin, it eventually ….. profits!
The actual pathway
Within step (2) of the pathway above lies the mystery of why each and every cell in the body can malfunction, either by consuming more glucose than it should (some cancers, some cases of obesity), or not accepting glucose when insulin is present (type 2 diabetes). The figure below illustrates how complex the pathway actually is.
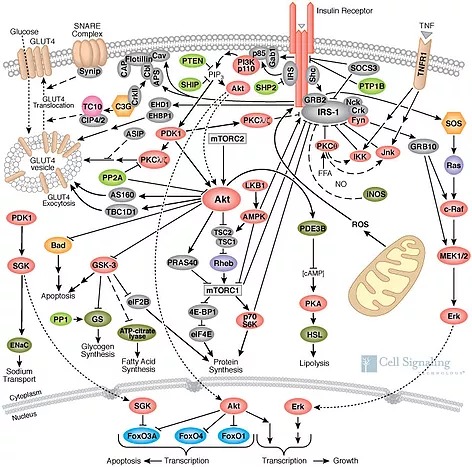
These numerous signaling molecules are effectively neurons in a cellular deep neural network. It should be no surprise that this is where all the cell “thinking” occurs, and it is these interactions that allow the cell to make the correct “decision” of whether or not it should accept additional energy when the body gives it “permission” to with the insulin hormone.
The full insulin signaling pathway remains incompletely charted, but we will describe the first few steps. After insulin binds to the insulin receptor (top orange) on the outside of the cellular membrane, the receptor becomes bound to another protein, the Insulin Receptor Substrate (IRS-1, gray) inside the cell. This is a fascinating protein that acts as a central hub of the whole pathway, but its primarily recognized function is to bind to and switch-on the protein known as PI3K (pink) and drag it to the membrane surface.
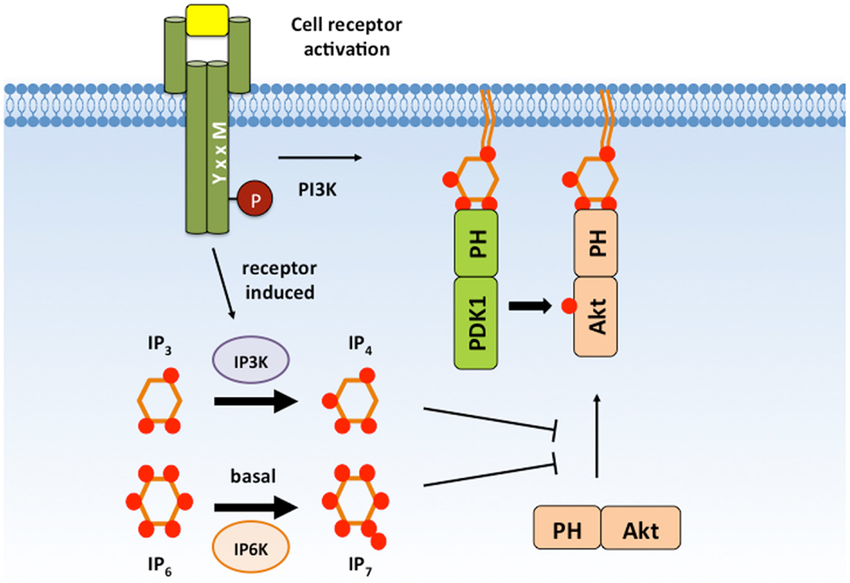
And this is where things magically come alive for this pathway. The PI3K enzyme modifies special lipid molecules in the cell membrane (PIP2 lipids) by adding an extra phosphate to them (making PIP3). When this happens, any free-floating protein within the cell that has a special adapter (PH domain) will randomly bind to these PIP3 molecules and become anchored to the inner surface of the cell membrane. When these molecules become confined to the membrane, they interact with eachother far more frequently. The cell membrane, being a confined 2D surface where signaling molecules gather to communicate, is effectively the “brain” of the cell. If all goes well, the cell will make the appropriate decision to let the right amount of glucose in. If the cell however is insane in the membrane, things can go wrong.
The PI3K enzyme recruits hundreds of signaling molecules to the membrane, and is a key activator of 1) cellular uptake of supplies and 2) growth of the cell. It shouldn’t surprise you then to learn that it is a top gene mutated in cancer (~40% of breast cancers), and its activity in muscle is decreased in diabetes. It is also a central focus of aging research. In fact, the PI3K gene is named AGE-1 in nematode worms because it was the first signaling pathway found to regulate lifespan! This pathway is the key link between aging/longevity science and human health.
The obesity link
In type 2 diabetes, skeletal muscle ignores the hormone insulin and refuses to accept glucose from the blood. This results in elevated blood glucose, much of which will be disposed of in fat tissue instead. For whatever reason, various signaling pathways present in step (2) block the insertion of glucose transporters in the membrane. Diabetes labs have a core focus on those signaling pathways, and have discovered strong links to inflammatory signaling through the tumor necrosis factor (TNF) pathway, which is an important signaling mechanism that allows the immune system to issue orders to other cells to shut down their metabolic imports. In the pathway map above, you can see the TNF alpha receptor (TNFR1) pictured to the right. When activated, its job is to switch off the early parts of the signal.
These inflammatory processes are well recognized in muscle in type 2 diabetes, but what about the highly related disease obesity? Fat cells have insulin receptors just like muscle cells do, and the proper functioning of these signals is required to keep the body healthy. Fat cells should only accept extra glucose from the body and store it, while at the same time leaving plenty for the rest of the body to use. The human body wants the fat cells at the back of the line, not the front! Could they potentially become deranged?
Greedy fat cells
At the turn of the 21st century, an Indian doctor Nikhil Dhurandhar made the important observation that viruses exist in animals that cause obesity. A virus known as SMAM-1 was discovered that infects chickens and causes them to immediately gain weight. Dr Dhurandhar hypothesized that similar viruses could exist that infect humans, and traveled to America to find out. In humans, it is not possible to ethically infect them with viruses for research, so he set out to study viruses known to infect humans to look for associations. Of the many viruses found in humans, the large family known as adenoviruses are highly prevalent and can cause the common cold. Most of us become infected with this family of viruses when we are young, and we fight them off.

Dr Dhurandhar began studying the known human adenoviruses, and discovered that Human Adenovirus #36 can infect mice, and demonstrated directly that infected mice gain large amounts of fat as a consequence. He then demonstrated that antibodies against this virus are easily found in the blood of Americans. In fact, more than %30 of obese people have antibodies against this virus, suggesting that they previously were infected and fought it off. Only %10 of lean individuals have antibodies to the virus.
Statistically, these numbers are quite significant, however the scientific field was not yet convinced of the causality. Many scientists argued the obese individuals and animals may just be more vulnerable to infections. In the last few years however, the actual molecular mechanism of adipogenic infections has been elucidated, directly proving a causal mechanism.
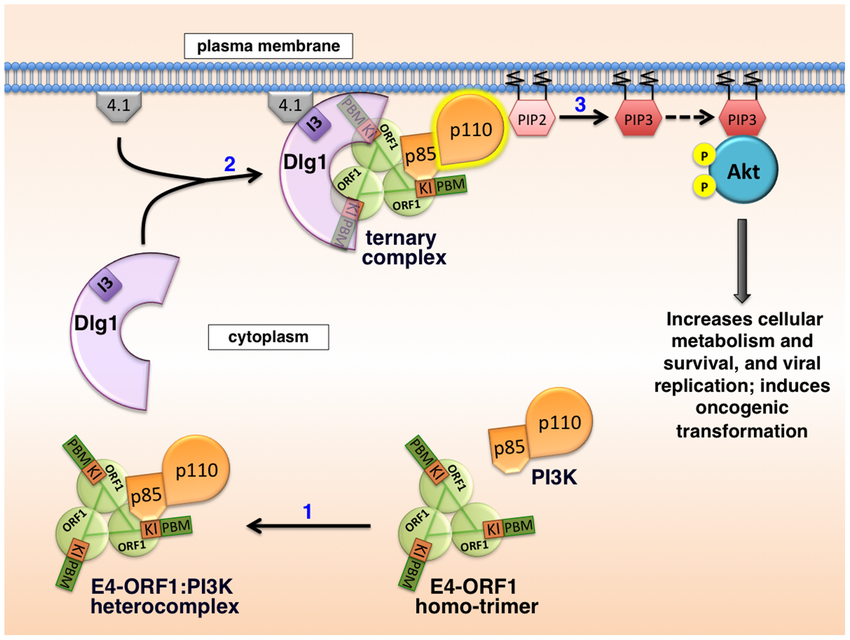
In 2014, Kong et al demonstrated that adnoviruses can, in fact, directly hijack cellular signaling. Through a common shared protein known as E4-ORF1, adenoviruses like the obesity-causing human Adenovirus #36 can trick an infected cell to import glucose even if it doesn’t want to. To accomplish this, the protein directly binds to the PI3K enzyme (via the subcomponents p85 and p110 pictured here) and activates it permanently. This activates the insulin signaling pathway without insulin being present!
The result of this sneaky trick is a greedy fat cell that robs the body of glucose. It skips to the front of the line entirely.
At the time of this writing, this exciting molecular data is only 5 years old, but I predict it is about to change everything we know about obesity and metabolic syndrome. The discovery of this molecular hijack of the insulin signaling pathway has provided strong evidence that these infections are causal of obesity, and not just correlations.
Everything you think about obesity is about to change
This is the part where I predict numerous research fields are about to collide with eachother. Buckle up. I’m either going to blow your mind here, or I’m just deluding myself. Made scientist time!

Obesity and nutrition
As you know, there is currently a substantial shift happening right now in nutritional dogma. The food pyramid has been turned upside down in recent years, fighting against the decades-held advice that high fat diets are harmful. Modern skeptics point out that the shift to high-carbohydrate diets are responsible for the obesity and diabetes epidemic.
I’m not taking sides here, but there might be some truth to this. I previously discussed genetic evidence that type-2 diabetes may have been largely caused by manmade chemicals, specifically compounds like BPA that suppress core signaling pathways that regulate the development of muscle and fat cells. It’s well recognized that obesity and diabetes are distinct symptoms with strong overlaps, and this infectobesity model presented here may be the key reason! When a fat cell becomes hijacked by a prevalent common-cold virus that infects more than 30% of us, it becomes greedy for glucose. When combined with environmental contaminants that cause skeletal muscle insulin resistance, there exists a “perfect storm” of high blood glucose to fuel infected fat cells!
What is not yet known is whether or not these common viral infections cause a preference for glucose over other energy sources, but if there IS a preference for glucose, this phenomenon may explain why low-carb diets allow many to lose weight. Maybe this is the only way to starve out the persistent infection! This hypothesis needs to be tested. (Seriously, our nonprofit needs funding)
Obesity and inflammation
I previously discussed the fact that the immune system has a central role in metabolic syndrome. It has long been known that the immune system invades muscle, fat, and liver tissue in obese and diabetic humans, where they can secrete pro-inflammatory signals like TNF-alpha. As discussed above, these signals allow the immune system to externally switch off a cells glucose uptake. In my prior writeup, I highlighted how genetically altering two key genes (Osteopontin and ACP5) can completely protect mice from gaining weight on a high-fat diet, or alternately produce mice that are extremely obese without any sign of diabetes.
The signaling pathways we found in human subjects at the Mayo clinic, and these prior mechanisms elucidated in lab animals, suggest that the immune system is a central player in metabolic system. I finished my PhD work in 2013, and was confused for years why macrophages would enter peripheral tissues and have the “final say” over that tissues energy usage. In light of these recent discoveries of viral infections however, the answer is obvious! The immune system has evolved to police our fat and muscle tissues looking for signs of infection. If macrophages become activated into the M1 proinflammatory state, they secrete signals that cause insulin resistance in muscle and fat cells by signaling through the TNF1R receptor, pictured above. This obesity-causing adenovirus36 however has found a trick around this defense however! By binding directly to PI3K, it can switch the glucose transport back on, effectively holding the cell hostage. Our immune systems haven’t found a way around this yet.
Obesity and aging/senescence
While clinicians continue to fight the very human obesity epidemic, biologists studying longevity and aging have focused on model organisms and cellular aging models in the lab. I’ve already pointed out significant overlaps between metabolic syndrome and whole-organism aging at the central insulin signaling axis, but the similarities are about to get more clear. I predict that the two field are about to collide in a profound way.
As previously discussed, a central focus of aging research is the phenomenon of cellular senescence, wherein a cell stops dividing for myriad reasons. As humans and animals age, the body becomes resident with these senescent cells. In 2010, the Campisi lab showed that senescent cells secrete a variety of cytokines into neighboring tissues that can effect the health and function of neighboring healthy cells. This was denoted the Senescence Associated Secretory Phenotype (SASP) and is thought to be partly responsible for the decline of organ function with age. This model lead to the hypothesis that removal of senescent cells may be beneficial. In 2011, the Van Deursen lab demonstrated that clearing senescent cells out of a genetically modified mouse will substantially improve its lifespan. The last decade has thus been a golden era for cellular aging and senescence research, showing quite clearly a causal mechanism between this phenotype and health declines with old age.
James Kirkland at the Mayo clinic has long argued that fat tissue in particular is a dominant site in which senescent cells accumulate. As humans age, fat tissue increases, and as senescent cells become resident in these fat deposits, inflammatory cytokines such at TNF-alpha and IL6 increase, leading to the proinflammatory state associated with obesity. I argued previously that cellular senescence largely resembles viral infections, primarily because both phenomena involve halting of the cell cycle in places that are metabolically favorable to viruses that are stealing energy for their own replication. Could senescent cells in adipose tissue be merely the result of viruses?
Obesity and genetics
Given that body mass index is easy to measure, it is no surprise that obesity is one of the most studied and understood human traits in the genomics literature. Studies of hundreds of thousands of people have mapped a handful of human genes associated with body mass index. Obesity is highly heritable, with studies showing that anywhere from 40%-70% of obesity can be explained with genetic factors alone. From the plot below, you can see that a few genes have very significant peaks, with many single genomic variants found in people that contribute to BMI. Many of these genes, in particular the 2nd most significant, MC4R, are found to be part of specific circuits in the brain that regulate feeding and metabolism. These genes and neural circuits will be discussed in a later post.
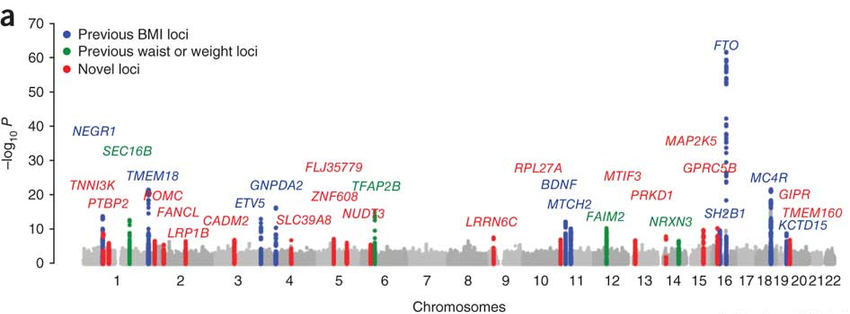
That top gene however, FTO, continues to confuse obesity researchers. Despite being the strongest genetic predictor of excess BMI, its role in the illness is far less clear. Unlike many of the other genes, it’s found throughout the whole body instead of being present preferentially in brain feeding circuits. It’s action in the cell is even more confusing.
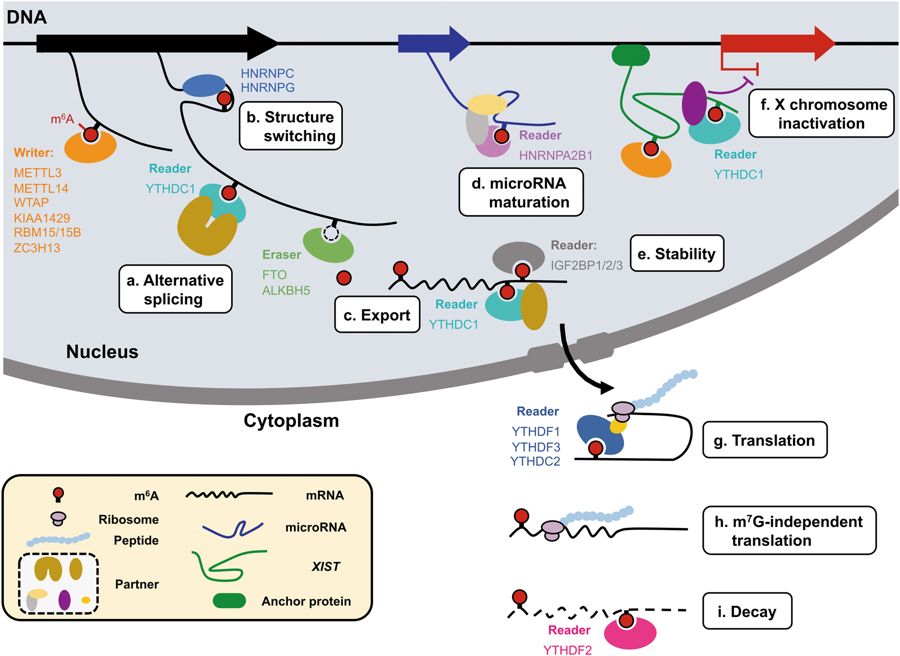
This enzyme resides inside the cell nucleus, and has a rather global influence on gene expression and translation. If you are familiar with the central dogma of molecular biology, you’ll know that genes in the genome are transcribed (copied) into mRNA “messages”, that are then exported out of the nucleus and converted into proteins through a process called translation.
In recent years, it has become apparent that mRNAs are heavily modified in the nucleus after they are transcribed. Among several new modifications discovered is adenosine methylation (m6A) that is found on some ~10% of the ‘A’ letters. These methyl groups are written, read, and erased in the nucleus, and the complex interaction of these newly-found pathways are the subject of the emerging science of “epitranscriptomics”.
These modifications add a whole new layer of cellular control of genes, just as epigenetics added a new layer of complexity onto genomics. Critically however, it is now known that the FTO gene, the top predictor of obesity in humans, is an eraser of these modifications. As you can imagine, this revelation has been confusing obesity biologists, since there are so many genes involved and regulated by this mechanism. If you spend any time investigating the mechanism by which FTO regulates feeding or metabolism, you will find a substantial number of theories of how it is regulating specific obesity genes, such as leptin, ghrellin, inflammatory cytokines such as TNF-alpha and IL6, and many others. Given that m6A modifications are found abundantly in gene transcripts, determining the precise causal connections to obesity has been impossible.
Future Predictions : A genetic risk model of viral infection
If you ignore the obesity literature for a time, and focus on virology, you will find that m6a modifications are known to have central roles in regulating the spread of viruses, and in fact, an emerging field of viral epitranscriptomics has recently emerged. In recent years, m6A modifications on mRNA has been shown to both accelerate and inhibit the spread of different viruses. The mRNA transcripts of genes from Adenovirus-2, for example, have about twice as many m6A modifications as cellular genes. Given that FTO is an enzyme that erases these m6A modifications, and that overexpression of FTO leads to obesity, it could be that these RNA modifications are serving as an antiviral mechanism.
The most exciting evidence I have found yet is this single grant application from a lab in the UK. Studying Kaposis’ Sarcoma HerpesVirus (KSHV), the lab of Adrian Whitehouse has discovered that the FTO gene has a critical role in the infectious process of this virus. Specifically, the virus has a specific protein that hijacks FTO and relocates it within the nucleolus, a special compartment within the cell nucleus that grows in size with aging. When this happens, it is prevented from “erasing” the mRNAs of the cells genes and instead removes methylation preferentially from the viruses genes instead. With this nasty trick, this virus effectively hijacks the cell and prioritizes its own replication. Most interestingly to me, KSHV is also known as Human HerpesVirus 8 (HHV8), a member of the gammaherpesvirus family, of which the very common Epstein Barr virus (HHV4) is a similar strain.
What happens next?
I predict that within the coming decade, we may find that these central cellular mechanisms of gene regulation, via the writing and reading of m6A modifications to specific genes, will be demonstrated to be a key mechanism behind the biology of obesity and aging. As part of this, it may be demonstrated that the FTO gene is similarly hijacked by adipogenic viruses such as Adenovirus36, present in more than 30% of obese Americans. I predict we will also discover similar infectious mechanisms in the brain from gammaherpes viruses that are known to have tropism for neurons.